AI, ML, and DataBiologySciencecomputational biologyDeepMindGoogleprotein foldingprotein structure
Just over a week ago the long-awaited AlphaFold2 (AF2) method paper and associated code finally came out, putting to rest questions that I and many others raised about public disclosure of AF2. Already, the code is being pushed in all sorts of interesting ways, and three days ago the companion paper and database were published, where … Continue reading →
Show full content
Just over a week ago the long-awaited AlphaFold2 (AF2) method paper and associated code finally came out, putting to rest questions that I and many others raised about public disclosure of AF2. Already, the code is being pushed in all sorts of interesting ways, and three days ago the companion paper and database were published, where AF2 was applied to the human proteome and 20 other model organisms. All in all I am very happy with how DeepMind handled this. I reviewed the papers and had some chance to mull over the AF2 model architecture during the past couple of months (it was humorous to see people suggest that the open sourcing of AF2 was in response to RoseTTAFold—it was in fact DeepMind’s plan well before RoseTTAFold was preprinted.) In this post I will summarize my main takeaways about what makes AF2 interesting or surprising. This post is not a high-level summary of AF2—for that I suggest reading the main text of the paper, which is a well-written high-level summary, or this blog post by Carlos Outeiral. In fact, I suggest that you read the paper, including the supplementary information (SI), before reading this post, as I am going to assume familiarity with the model. My focus here is really on technical aspects of the architecture, with an eye toward generalizable lessons that can be applied to other molecular problems.
For AlphaFold2, the apparent answer that DeepMind gave to the question of what they should do is… yes. Self-supervision? Yes. Self-distillation? Yes. New loss function? Yes. 3D refinement? Yes. Recycling after refinement? Yes. Refinement after recycling? Yes. Templates? Yes. Full MSAs? Yes. Tied-weights? Yes. Non-tied weights? Yes. Attention over nodes? Yes. Attention over edges? Yes. Attention over coordinates? Yes. The answer, to all the questions, is yes! And this clearly paid off.
My somewhat flippant characterization may give the impression that AF2 is a mere smorgasbord of good ideas—this couldn’t be further from the truth. AF2 not only includes every conceivable feature but integrates these features in a remarkably unified and cohesive manner. It is as if all the team’s disparate ideas were repeatedly fed through the same intellectual bottleneck so that they emerge homogenized (their recycling approach, applied to the AF2 design process itself.) The result is both a tour de force of technical innovation and a beautifully designed learning machine, easily containing the equivalent of six or seven solid ML papers but somehow functioning as a single force of nature.
One sentence in the main text is particularly telling, as each phrase essentially corresponds to a whole paper, one of which has already been reported by a team at Facebook Research as a separate and impressive effort in its own right (the MSA Transformer; by all indications developed contemporaneously.)
“… we demonstrate a new architecture to jointly embed multiple sequence alignments (MSAs) and pairwise features, a new output representation and associated loss which enable accurate end-to-end structure prediction, a new equivariant attention architecture, use of intermediate losses to achieve iterative refinement of predictions, masked MSA loss to jointly train with structure, learning from unlabelled protein sequences using self-distillation, and self-estimates of accuracy.”
Suffice it to say that I am more impressed with AlphaFold2 after having read the paper than before. This is a work of art, one with numerous conceptual innovations that have nothing to do with compute power or “engineering” and is a more intricate tapestry than I had anticipated after the initial details were released. Soon after CASP14, I had a discussion with a friend in which the topic of how far the academic community was behind DeepMind came up. At the time I said it would have likely taken ten years of academic research to achieve what they did, while he thought it was more like two. After he said so I backtracked, thinking that perhaps I was being harsh. Now that I have read the paper, I think it would have likely taken at least 5-6 years before the academic community’s effort could have added up to AlphaFold2. (On this point, the right delta to measure is not RoseTTAFold vs. trRosetta, but trRosetta at CASP14 vs. Rosetta at CASP13; even then the gap is in large part due to the first AlphaFold.)
What I will do here is make a series of observations that summarize, from my perspective, the most interesting and surprising aspects of this unified architecture. They are (somewhat) listed in my perceived order of their importance.
Disclaimer: everything I say below is based on my understanding of AlphaFold2 and how it might be working, which may obviously be incorrect. It is a complex system with many moving parts, and I have no additional insights beyond what is provided in the Nature papers.
The most important line in the whole paper, IMO, is line 10 of algorithm 20 in the SI, particularly when combined with the ablation of Invariant point attention (IPA) in Figure 4a in the main text, the core component of the structure module. If there is one takeaway for me, it is this line, as in it I think lies the crux of the “magic” of AF2. I say this not because of the line itself per se, but because of what it implies.
Let me back up a minute.
One of the challenges of building ML systems that reason over proteins is the fact that proteins are long 1D polymers that fold in 3D space. Key here are “1D”, “3D”, and “long”. On the one hand, given the sequential nature of proteins, it is natural to encode them using sequence-based architectures. When I developed the RGN model I did just that, using what was at the time the leading architecture for sequence problems (LSTMs). However, architectures of this sort have trouble reasoning over long-range interactions, a common phenomenon in proteins. Still, 1D representations are convenient because they readily map to the physical object, permitting parameterizations (e.g., internal coordinates) for predicting protein structure in a straight-forwardly self-consistent manner.
Most of the field, up to and including the first AlphaFold, pursued a different approach that relies on 2D matrices to encode structure using inter-residue distances. This captured long-range interactions better (relative to sequential models) but introduced the awkwardness of mapping a 2D representation to an object that is fundamentally a 1D curve in 3D space. Typically this mapping was accomplished using physics-based relaxation or SGD (the first AlphaFold) in a post-processing step, but left much to be desired.
When DeepMind first revealed AF2 at CASP14, they described what appeared to be a hybrid 2D/3D approach, in which the structure is initially encoded in a 2D representation that is then transformed into a 3D representation. There was much speculation about how this was done (more on this later), but it left, in mind, this awkwardness about the 1D-2D mismatch. This is a subtle point. The question here is not about how to map a 2D distance matrix to a 3D set of coordinates (many approaches exist.) Instead, it is about the structural mismatch (in terms of data / tensor types, not protein structure) between an object that is fundamentally one-dimensional (the protein itself) and its distributed representation in a 2D matrix. One can come up with various hack in terms of averaging rows and columns to transform the 2D representation into a 1D one, but they always struck me as just that, hacks.
What I did not anticipate, and what I find quite elegant about the AF2 architecture, is the central role that the MSA representation, and in particular the first row of that representation—which initially encodes the raw input sequence but is ultimately projected onto an object they denote —plays in the AF2 symphony. In some fundamental sense, the central encoding of a protein in AF2 is wholly one-dimensional—it is this 1D object, , that ultimately captures what is needed to predict the 3D structure. This is where line 10 of algorithm 20 comes in. From that humble , the entire structure is projected onto 3D space, without any kind of explicit 3D reasoning. It is true that in the full AF2 model, the IPA module does operate in 3D and benefits from one-way (incoming) communication from the 2D representation, but, remarkably, when this module is ablated, AF2’s performance remains nearly unchanged from the full version (so long as recycling is enabled—more on this too later.)
This was the most mind-blowing aspect of the paper to me, the part that felt most “magical”. And the way they’ve constructed it is very clever. It is indeed the case that learning using a 1D representation is difficult, especially for long-range interactions. So what AF2 does is use a 1D object to represent the protein, but couple it to a 2D object (the pairwise ) to overcome the shortcomings of the 1D representation. I suspect that implicitly acts like memory for , helping it to store information that it can reference and iterate on during training and prediction (I don’t literally mean , as that is the last projected representation, but I’m overloading notation here—I actually mean , the first row of the MSA representation in the evoformer.) More broadly, likely facilitates the learning process, by providing a richer representation for the model to work with. At every step of the process, is kept updated, communicating back and forth with , so that whatever is built up in is made accessible to . As a result is front and center in all the major modules. And at the end, in the structure module, it is ultimately , not , that encodes the structure (where the quaternions get extracted to generate the structure). This avoids the awkwardness of having to project the 2D representation onto 3D space.
While I was reading the initial manuscript and making my way to algorithm 20, I felt rapt in anticipation of how the structure will finally be extracted. When I discovered that it was and not that truly mattered, I felt a sense of genuine giddiness.
In a way what the AF2 team has done, if I am allowed to speculate here a little, is to develop an approach where the representation used for learning and reasoning is decoupled from the representation used to hold the state of the system and predict structure, yielding the best of both worlds. In most ML architectures, a single representation is used to do both; in AF2, it is split. When DeepMind first announced their hybrid pair/MSA representation, I didn’t really get the “point” of it, but now that I understand it in detail, I suspect the above was one of their key motivations, although of course I have no way of knowing for sure.
Side note: when they incorporate structural templates, they add torsion angles as rows to (again actually ), and so structural information is being explicitly embedded in early on. Of course, templates are not guaranteed to be available during inference time, but their occasional availability during training means that the evoformer has to learn how to reason with this type of geometric information in from the very beginning.
Information flow is key
If there is one unifying theme to the paper, one broad principle, it is that AF2 is engineered to maximize information flow between its components (all greased by liberal but intentional use of ). Furthermore, it does so in a way that reflects aspects of our understanding of proteins. I was tempted to put this first on the list but opted not to because it is less of a concrete idea or trick and more of a prevailing characteristic of the architecture.
I will illustrate this with a couple of concrete examples. First is the communication between the pair representation and the MSA representation (2D and 1D from before, although the MSA representation is actually 2D too, over sequences.) At each iteration, both update one another, ensuring constant synchronization. The updates are asymmetric, however. The MSA → pair direction is rather heavy duty, using the module to update every component of . This, again, is perhaps reflective of the central role that ultimately plays (recall that is basically ). However, the MSA representation is not uninfluenced by the pair representation. The latter updates the former as well, but through a light touch, biasing how different columns of the MSA representation attend to one another. The fact that it is a light touch, instead of the heavy-duty cross-attention mechanism that I at least had expected, may or may not have been done for computational expedience. However, the fact that influences the column-wise attention of makes perfect biological sense, because the pair representation ought to encode coupling strengths between residue positions. This also avoids the “hack” I mentioned earlier, of having to average rows or columns to extract 1D information—it is never done, because the individual elements of directly bias the individual attention elements of (lines 3 and 5 of algorithm 7.)
Another example is the communication within the pair representation, which utilizes two forms of novel “triangular” attention performed between pairs of pairs of residues—naively a very expensive computational operation. Here too efficiency is achieved by using a light touch approach of only biasing the attention, and more importantly by restricting attention to pairs of pairs that have one residue in common, with the intuition being that the triangular inequality ought to constrain such quantities (because three residues form a triangle.) This one is interesting because it illustrates an important principle: geometric constraints need not be realized literally, as many people, including my group, had been trying to do, by e.g., mathematically enforcing the triangular inequality, but instead informationally, i.e., in the information flow patterns of the attention mechanism. In effect, what AF2 does is convert geometric intuition—we know these two pairs of distances ought to constrain one another—into architectural features that allow these pairs of distances (or rather the representations encoding them) to communicate easily. This to me is a general principle of ML architectural engineering, particularly for problems with rich domain knowledge. It illustrates how prior information ought to be integrated into learnable models: not through literal hard constraints or even soft ones, but as aspects of the learning process itself. (I would say this is broadly true of deep learning, vis-à-vis say probabilistic programming or Bayesian modeling. Knowledge is rarely injected directly in DL; it is instead infused into the architecture through designs that make it easier to learn. In this sense, hard SE(3)-equivariance is actually an exception, but more on that later.)
One last example is in the IPA structure module, which when enabled gets both a light touch from the pair representation in line 7 of algorithm 22, where biases attention between residues in 3D space, as well as a heavy touch in lines 8 and 11, where is made directly available to the 3D reasoning engine. The light touch again makes perfect sense, as the pair representation should encode which residues are spatially close, a valuable piece of information when reasoning spatially.
Crops are all you need
AF2 (and the first AF before it, although not quite in the same way or extent) is trained in a seemingly strange way: not on entire proteins, but on fragments of ones, or what the AF2 team calls ‘crops’. They are not short; usually a couple of hundred residues. But for longer proteins, these crops only capture a small fraction of the whole sequence. Furthermore, two non-contiguous crops are often made (from the same protein), and then stitched together. Remarkably, while AF2 is mostly trained on crops of up to 256 residues (later fine-tuned on 384), it can predict protein structures with well over 2,000 residues, an astonishing feat. Not only because it is a very hard problem in absolute terms, but also because AF2 is trained on much shorter crops.
Global context matters in proteins. The same subsequence of amino acids, in two otherwise different proteins, will not in general have the same structure; protein structure prediction would have otherwise been solved long ago! (Of course, this gets progressively less true as the length of the subsequence grows.) Taking disembodied crops, that can be hundreds of residues apart, and asking the model to predict their relative orientation when even the length of their separation is unknown (if they are more than 32 residues apart) seems like an impossible task. But there are at least two ameliorating factors at work here. First, AF2 is working with MSAs / co-evolution patterns, which encode this information irrespective of linear chain separation. Second, this is only done at training and not inference time. During inference, AF2 does have access to the whole sequence, and so the issue of context-sensitivity is moot. During training, AF2 may get one signal from one pair of crops in one protein, and a conflicting signal from a similar pair of crops in a different protein, and that would be totally fine. Such situations likely teach the model that there is inherent uncertainty when it sees these two crops at some unknown separation. It is being taught the context-sensitivity of proteins. Recall that, during training, there is no requirement that the model is ever able to accurately predict the structure. All that matters is that useful information is being imparted to the model through gradient updates.
This is another instance of decoupling usually coupled things (the previous one being suitable representations for learning and holding state.) In most ML models, the training and inference tasks are kept very similar, with the idea that the more similar the training task is to the inference one, the better. But this is not really necessary, given that training is about acquiring useful signal from the data while inference is about making accurate predictions. This idea is certainly not unique to AF2; generative models often involve quite different tasks during generation vs. training, and of course differentiable loss functions used during training are often different from the true target functions. Still, AF2 demonstrates a rather robust use of this idea in the supervised learning context, almost arguing for intentionally decoupling tasks, when in general people have tended to treat such decouplings as failures of modeling.
In fact, it appears that the architects of AF2 had this in mind as a design feature, an inductive prior for the model. This shows up most visibly in the relative positional encoding of residue pairs, which are capped at 32 residues apart (i.e., in terms of raw inputs, if two positions are more than 32 residues apart, they are treated as 32 residues apart.) For contiguous crops, this is not really a limitation because the model can learn to add positional embeddings so that it can infer the true separation. But for non-contiguous crops, it will have no idea what the separation distance is. Yet, the model is being tasked with figuring out how the two crops should be oriented with respect to one another. This is an inductive prior that states that, in protein-land, if two crops are beyond a certain distance apart, it doesn’t really matter how far apart they are exactly. It is an interesting prior, and one that seems to have paid off for AF2. [Update 7/27/21: Turns out that the multiple non-contiguous crops scheme was a feature of the manuscript I reviewed but not the final version, as it was not used in the CASP14 model of AF2.]
I don’t know if the above was the intention for the AF2 team going in—memory efficiency likely played a big role in their thinking, given the near impossibility of training something as large as AF2 on full-length proteins using current TPUs. Nonetheless, what may have started out as a computational trick ended up being a good idea biophysically-speaking.
Incidentally, this resolves one of the great mysteries about AF2 during CASP14—the gap between inference and training times. Because of cubic scaling, and the fact that inference is done on full-length proteins while training is done on crops, there (can be) a very large compute-time gap between the two.
Always a refiner
Another striking feature of AF2 is its always-on refinement mode. I.e., it is always capable of taking a preliminary structure at some distance from the native state and refining it to be closer to that native state. This is true in multiple modules and at multiple levels of granularity, making the system remarkably robust and versatile at utilizing diverse types of data.
It is most obvious and natural in the structure module, where the weights of the iterative IPA procedure are tied and so the same operations are applied repeatedly. This makes sense as IPA’s intended function is to refine the structure coming out of the evoformer. However, the evoformer itself is also always in refinement mode. This is not explicitly encoded in the architecture per se (the weights of the 48 layers of the evoformer are untied) but is evident in the way it is trained, where it is encouraged to behave this way. For example, the raw inputs can include templates of homologous structures, some of which may be similar to the sought structure, thus providing the very first layer of the evoformer a structure (encoded in the pair representation) that is essentially complete and that should not be screwed up. This is key and encapsulates what is hard about this, because AF2 may also get a sequence that has no structural homologs, thus providing the first layer of the evoformer with virtually no structural data—in both instances, the evoformer must learn to behave correctly. Repeated subsampling of the MSAs reinforces this, because each sample provides varying degrees of sequence coverage.
The same phenomenon also occurs with recycling. First, recycling itself is a form of refinement, as the entire network, with tied weights, is reapplied up to three additional times. But the act of recycling also teaches the evoformer to be a refiner, because the same evoformer in a later recycling iteration can be presented with structures that are much further along than the evoformer in the first (pre-recycling) iteration.
Another mechanism to encourage refinement is the use of intermediate losses, both in the structure module and in recycling. I.e., during training the model is optimized to minimize losses of the final predicted structure as well as those of intermediate structures predicted some fraction of the way through the system. This encourages AF2 to not only predict structures correctly, but to do so quickly, in earlier iterations of the system. In the structure module this is done very explicitly; its loss function is literally an average over all iterations. In recycling it is a bit more subtle. The loss from only one iteration is used for backpropagation, but because the number of iterations is stochastically sampled, the effect is the same; the model is encouraged to get structures right in earlier recycling iterations.
AF2’s robustness to varying tasks at varying stages is evident in the animations supplied with the paper. The video of LmrP (T1024) shows a structure that is essentially complete after the very first layer of the evoformer, while that of Orf8 (T1064) goes on and on till the very end, almost resembling a molecular dynamics folding simulation (it is not one, obviously.) Incidentally, these animations are also suggestive of AF2’s behavior with respect to sequences of varying MSA depths. For deep MSAs, it perhaps acts similarly to pre-AF2 methods that relied heavily on co-evolutionary signal, inferring the structure more or less entirely based on that signal with just a single evoformer layer. For sequences with very shallow MSAs, it falls to the later stages of the evoformer and structure modules to actually fold the protein, where I suspect the model is learning and applying general physics knowledge about protein structure. The “No IPA and no recycling” panel of Supplementary Figure 5, which shows AF2’s performance degrading substantially for shallow MSAs when recycling and IPA are turned off, supports this hypothesis. Furthermore, AF2’s apparent success in predicting proteins complexes from unpaired MSAs may be due to this general physics knowledge, although Sergey Ovchinnikov has a compelling alternate theory.
On the whole I find the idea of constant refinement powerful and broadly useful, especially when it can be applied without having to backpropagate through the entire model. I am not sure if this is novel within the iterative supervised learning context (it probably has parallels in generative computer vision). It is very RL-like, which is obviously DeepMind’s forte.
The why of SE(3)-equivariant reasoning
Possibly my biggest surprise reading the paper occurred when I came across the ablation studies of Figure 4a, particularly the ablation of IPA, AF2’s much ballyhooed SE(3)-equivariant Transformer. While I did not ascribe as much value to this module as I think others did pre-publication, the fact that removing it seemed to do so little was still shocking. What was the point of all this work and machinery if it contributed so little?
There were actually two surprises here. First is the fact that without IPA, AF2 simply spits out 3D coordinates, without any explicitly SE(3)-invariant conversion of the “distances” in the pair representation to 3D space. In fact, as I mentioned earlier, the IPA-less version of AF2 relies entirely on the 1D for structure generation. This means it picks a specific global reference frame in which it generates the structure, which I think to many people, myself included, seemed too crude to work. But apparently it does just fine.
The second surprise is the fact that reasoning in 3D, i.e., reasoning after an initial version of the structure is materialized in a global reference frame, appears to not be terribly important, unless recycling is also removed. This flies in the face of our intuition that certain spatial patterns, particularly ones distributed across multiple discontiguous elements of the protein, are more readily apparent in 3D space, and should therefore benefit from 3D reasoning. From a practical standpoint, it also seems to obviate all the methodological research that has gone into equivariant networks, at least insofar as it applied to proteins (more on this in the next section.)
This is certainly one interpretation, but I don’t consider it to be an entirely accurate one. The key lies in the fact that removing IPA is ok only as long as recycling is retained. When both are ablated, as Figure 4a shows, performance drops considerably. Furthermore, if recycling is removed but IPA is retained, then AF2’s performance stays nearly unperturbed. This is a rather impressive showing for IPA, given that recycling is essentially quadrupling the 48-layer evoformer (computation cost-wise it is not, because of various tricks), while IPA is only 8 weight-tied layers. Viewed in this light, IPA layers are far more efficient than evoformer layers, at least for late-stage refinement. As the focus now shifts from single chain prediction to complexes and higher-order assemblies, the importance of spatial reasoning will only increase, and I expect that IPA and its future derivatives will continue to play an important role.
The how of SE(3)-equivariant reasoning
Setting aside the utility of SE(3)-equivariance, the question of how it is performed in AF2 was probably the most anticipated one prior to the publication of the paper. It helps to step back for a moment and consider where this subfield has been going for the last few years. The flurry of recent activity in equivariant neural networks arguably got started with a paper from Thomas et al., although there were some antecedent works. The paper relied on group-theoretic machinery, employing (convolutional) filters that used spherical harmonics as their basis set. The formulation is mathematically elegant and has been elaborated in numerous papers since, with one in particular by Fuchs et al., the SE(3)-equivariant Transformer, that not only generalized the approach from convolutions to self-attention but also shared an identical name to that used by the DeepMind team during CASP14 to describe what they now call IPA. This naturally led to the speculation that AF2 used something very similar to this approach, including in my own previous post on AF2. This, in retrospect, had little merit, especially since the approaches were developed around the same time and so there was no reason to believe they influenced each other.
Parallel to the development of group-theoretic approaches, there has also been a flurry of graph-theoretic approaches to the problem of reasoning equivariantly over molecules. Instead of relying on spherical harmonics, the graph-based approaches embed molecules as graphs, with spatial information encoded in the edges that connect nodes that in turn encode atoms. This line of research has been applied both to small molecules and to proteins, but arguably it is in the latter where it has found the most utility. Proteins, like all polymers, inherently permit the construction of unambiguous reference frames at each atom, and this fact has been exploited to great effect by graph-based approaches. One of the first—perhaps the very first—method to use this construction in the context of machine learning and proteins is the Structured Transformer by Ingraham et al., and it appears that this work was the inspiration for IPA. There are many advantages to using graph-based constructions over group-based ones for proteins, but this is a longer discussion that does not impinge on how IPA works. Suffice it to say that IPA falls squarely in the graph-based camp, and for proteins this IMO makes the most sense.
How IPA works is quite interesting, as it is arguably the most novel neural primitive in the whole of AF2, combining multiple spatial reasoning mechanisms that will likely inform much of future molecular modeling. I will focus on a few. I should emphasize that most of my thoughts are highly speculative, especially the ones on meta-reasoning.
First is the IPA attention mechanism, which is something of a beast (line 7 in algorithm 22). It includes the usual non-geometric query/key matching (). It also includes a bias term from the pair representation (), which is one place where my earlier point about “all roads lead to ” is untrue, but only if IPA is turned on. And finally, the most interesting piece, is the inclusion of geometric query/key matching based on 3D vectors “sprayed” by each residue (). In general, IPA does a lot of this “spraying”, where residues generate many 3D vectors both to control attention and to send values to one another. These vectors are generated in the local reference frame of each residue and then transformed to the global reference frame, so that they are both equivariant and able to attend to the entire protein in 3D space. Because the geometric matching term is negative, it attenuates rather than increases attention. This makes it rather conservative—leading most residue pairs to ignore one another—because it requires that the query/key are matched just right to avoid attenuation, which is unlikely to occur by chance. The degree of attenuation is a learnable per-head parameter (), and it is possible for IPA to learn to turn it off, but geometric matching can never positively contribute to attention. Taken together these aspects likely induce a spatial locality bias—it is easier for each residue to spray generic local vectors for the query/key, which when transformed to the global reference frame would attend to nearby objects, instead of a precisely positioned vector far away from a residue’s center, that when transformed to the global reference frame lands on exactly the right part of the protein.
Second is how values, specifically geometric values in the form of 3D vectors, are communicated between residues (lines 3 and 10 of algorithm 22.) Each residue again sprays multiple vectors, all transformed to the global reference frame, where for any receiving residue, the vectors of all other residues are averaged in Euclidean space (weighted by attention and done per head/point value) over the whole protein before they are transformed back to the local reference frame of the receiving residue. I imagine this enables sophisticated geometric reasoning capabilities, ones that may reflect general aspects of protein biophysics and are less tied to the genetic / co-evolutionary information contained in MSAs.
Consider for instance a “catalytic triad” in which one residue must detect how two other residues are positioned and oriented with respect to itself. It sprays query vectors with the appropriate positions and orientations, and because they are specified in its local reference frame, they form a generic spatial pattern that IPA can learn and apply repeatedly. If the query vectors are pointing in the right direction, we can imagine key vectors that simply state the location and orientation of each residue, essentially returning . When transformed to the global reference frame, the keys would match the queries, sparing attention from attenuation. Most other attention pairs would get attenuated; noise filtered. Now that the relevant residues are attending to one another, each returns a value that again encodes . This information is subsequently processed in line 10 and downstream layers to nudge the residues to align better with the IPA’s expectation of an active catalytic site.
Whether the above is an accurate description of how IPA works is of course unknown, and may not be knowable unless one carefully inspects the behavior of the learned weights and model.
Third, and by far most speculatively, is the possibility that IPA may combine its components to perform meta-reasoning. By this I mean reasoning not about protein structure, but about AF2 itself, namely the status of its knowledge about the current inference task and how it might be improved in subsequent IPA iterations (I am referring to learning in real-time during inference, not through gradient descent.) Imagine that AF2 expects one protein fragment to interact with another but is uncertain of its location. During the first iteration, it sprays multiple broadly spaced query vectors, distributed over a large section of the protein region that it thinks may contain the sought fragment. Nearly all queries won’t find a match, but one of them, if AF2’s hypothesis is correct, might. Once found and based on the information obtained, AF2 can in subsequent IPA iterations send more directed queries to better localize the exact position and orientation of the fragment, and then refine the structure according to its learned knowledge of proteins.
The key point here is the use of the iterative aspects of IPA to control its reasoning and discovery process. Compared to traditional sampling approaches, where different protein conformations are randomly considered, IPA (and the evoformer) may actively reason about how to improve its knowledge about the protein it is currently trying to fold. To be sure, there is no guarantee that this sort of meta-reasoning is happening, and I suspect it would be non-trivial even for DeepMind to assess this. If meta-reasoning is happening, it may explain the long folding times for Orf8 and other structures with shallow MSAs (in the sense of arriving at the native state.) If AF2 sprays space with vectors to find and orient protein fragments, it would be doing a form of search, one that can take many iterations to conclude. This would explain why the same computation, repeatedly applied, can eventually fold the protein, and the specific importance of IPA and recycling to proteins with shallow MSAs in the ablation studies of Supplementary Figure 10.
One last point: it is remarkable to me how, again, in line 10 of algorithm 22, all this geometric information is encoded back in , an object that is not explicitly geometric but all “latent”. Many equivariant architectures have focused on maintaining the explicitly geometric aspects of their representation throughout the network, but in IPA, explicit geometry is used only briefly, to perform the operations I describe above, before it is piped into and all is forgotten. It demonstrates both the power of encoding geometric information in and the advantage of relying on implicit representations that avoid the complications of formally maintaining equivariance throughout the network (in general, equivariant layers have a hard time playing nice with neural network nonlinearities, particularly the group-theoretic formulations, if not mathematically then at least from an optimization standpoint.)
MSA Transformer
This is a minor point methodologically but an important one performance-wise. AF2 includes the MSA Transformer as one of its auxiliary losses, and Supplementary Figure 10 shows it to be critical for sequences with shallow MSAs, where performance degrades substantially without it. To me, one of the most impressive aspects of AF2 is its robustness to shallow MSAs. Self-supervised learning over sequences (or MSAs in this case) has long seemed like a natural way to tackle the problem, and it pans out here.
Interestingly, this gain evaporates when the MSA is very shallow, e.g., just a single sequence, but this makes sense too because then the MSA Transformer is uninformative.
DeepMind’s magic is not in brute forcing scale
I will end with perhaps the most counterintuitive conclusion I took from the paper. Going in, I had anticipated that at least for some components, most notably the evoformer attention mechanisms which, without simplifications like axial attention, would scale rather horribly, DeepMind had exercised their enormous computing resources to brute-force their way through problems that other groups would have to be clever about. The irony is that the truth appears to be almost exactly the opposite. What is impressive about the AF2 effort is not top-notch hardware, but top-notch software and ML engineering that renders brute-force scaling unnecessary. This part I suspect will be hardest for academia to replicate, because it is less about increased national investment in computing resources (which would undoubtedly help) and more about in-house professional software engineering capacity, a much taller order.
This is evident in numerous places: the careful use of gradient stopping, in IPA but also in recycling, where an enormous amount of compute is saved; in cropping, probably the most important and impressive, because it so fundamentally changes the inductive bias of the system; and in thoughtfully chosen initialization schemes. It is true of course that DeepMind can have their cake and eat it too. Their use of 8 ensembles for CASP14 demonstrates this, where they increased compute requirements by an order-of-magnitude for what appears to be very marginal gains (I suspect they only discovered this after CASP14 however.) But on the whole, their culture appears to be one of computational frugality, in the best sense of the word, and quite possibly the biggest compliment I can give the AF2 team on an achievement that is remarkable in so many other ways.
AI, ML, and DataBiologyConferencesScienceCASPCASP14deep learningDeepMindGoogleprotein foldingprotein structure
The past week was a momentous occasion for protein structure prediction, structural biology at large, and in due time, may prove to be so for the whole of life sciences. CASP14, the conference for the biennial competition for the prediction of protein structure from sequence, took place virtually over multiple remote working platforms. DeepMind, Google’s … Continue reading →
Show full content
The past week was a momentous occasion for protein structure prediction, structural biology at large, and in due time, may prove to be so for the whole of life sciences. CASP14, the conference for the biennial competition for the prediction of protein structure from sequence, took place virtually over multiple remote working platforms. DeepMind, Google’s premier AI research group, entered the competition as they did the previous time, when they upended expectations of what an industrial research lab can do. The outcome this time was very, very different however. At CASP13 DeepMind made an impressive showing with AlphaFold but was ultimately within the bounds of the usual expectations of academic progress, albeit at an accelerated rate. At CASP14 DeepMind produced an advance so thorough it compelled CASP organizers to declare the protein structure prediction problem for single protein chains to be solved. In my read of most CASP14 attendees (virtual as it was), I sense that this was the conclusion of the majority. It certainly is my conclusion as well.
In a twist of irony the community most directly affected by this development—in some ways negatively affected on a personal level as AlphaFold2 (AF2) essentially obsoletes at least parts of our research programs—has been the most unanimous in its agreement on the significance of AF2’s advance (although certainly not wholly unanimous). Judging by Twitter, communities further and further away from protein structure prediction have had more mixed reactions.
In this post I will try to distill my views on AF2 and CASP14. I struggled with whether I should write this blog post as it felt at times like an obligation rather than something I desired to do. Sequels are also never as good as the original and the weight of expectation from my CASP13 post fazed me. In the end however there were enough new things to say that I felt it worthwhile to write the post. I hope that it proves useful to others.
In “The Advance” I explain the magnitude of AF2’s leap in quantitative terms; “A Solution?” addresses the controversy and semantics of the word “solution”, and unpacks the myriad problems often called “protein folding” but that are not; in “The Method” I speculate on what AF2 does exactly, although details here are thin because we don’t have them; “Impact on …” details my views on how this impacts fields ranging from protein structure prediction to the whole of biology; “Why DeepMind?” delves, a little bit, into the “sociology” of why it was DeepMind that managed this and not anyone else, although this section is not nearly as long as the one I wrote for CASP13 for the simple reason that most of what I said then still holds true IMO; and finally, in “Academic Research in a New Age” I put on my academician’s hat and think out loud about how best to compete strategically in this new era—this section is inside baseball for academics working on biomolecular machine learning and can be safely ignored by most.
Before I start (sorry) one last bit of housekeeping: after publishing my CASP13 blog post two years ago I received an entirely undeserved amount of attention and found myself repeatedly the spokesperson for a field far too big and diverse for me to have any right to represent. There are many people at least as capable as me and often more so in this space; among the “new” generation I count Sergey Ovchinnikov, John Ingraham, Possu Huang and many others. Among the “established” generation there is of course David Baker, Debbie Marks, Jinbo Xu, Chris Sander, Yang Zhang, and many more. And then there is the AlphaFold team. I only list people I personally know and have interacted with, but a quick perusal of the CASP14 participants list can give you an idea of who is active in this space. All of them are just as and often far more qualified than I am to speak about this problem and so please ensure that you get a broad representation of views when seeking to form your own.
To understand AF2’s significance and why so many people were yelling “OMG PROTEIN FOLDING IS SOLVED!!!!!” it makes sense to first take stock of the qualitative magnitude of AF2’s leap. I was privy to the results before they became widely known and my initial expectation when I heard that DeepMind will declare the problem solved was that they had achieved a median GDT_TS of around 80. You can intuitively think of GDT_TS (definition) as the fraction of the protein that is correctly predicted, i.e., 80 corresponds to ~80% of the protein being more or less right. Random predictions give around 20; getting the gross topology right gets one to ~50; accurate topology is usually around 70; and when all the little bits and pieces, including side-chain conformations, are correct, GDT_TS begins to climb above 90. I speculated in my CASP13 post that in ~4 years’ time we would get the topology correctly, i.e., we’d have a median GDT_TS between 70 and 90 at CASP15. Although I never wrote it in the CASP13 post, my expectation (back then) for when we would nail all the details was another 10 years, i.e., not until the late 2020s would we see >90 GDT_TS for most targets.
So when I learned that DeepMind will declare the problem solved I assumed they had done what they did at CASP13 again, achieving in two years what I thought they would do in four, getting a median GDT_TS of ~80. I was impressed and eager to hear the details. Imagine my surprise then when I was informed a few days later of the final number, a median GDT_TS of 92.4. Never in my life had I expected to see a scientific advance so rapid.
Statements have been made that this is proteins’ “ImageNet moment” but that would be incorrect IMO. The ImageNet moment was the first time deep learning demonstrated it can outperform conventional approaches on image recognition and made the field of computer vision take notice. Relative to AF2’s advance this year, the 2012 ImageNet advance was incremental. The closest to an ImageNet moment this field has had is Jinbo Xu’s 2016 PLoS Comp Bio paper, which demonstrated the first real impact of deep learning on protein structure prediction. This on the other hand is something altogether different. It is more akin to having the ImageNet accuracies of 2020 in 2012! A seismic and unprecedented shift so profound it literally turns a field upside down over night.
The table of Z-scores that CASP14 publishes has been making the rounds and so I won’t reproduce it here, in part because I think it’s a little hard to interpret other than saying “AF2 does much better than anyone else”. Instead, consider the graph below, which illustrates the delta between AF2 and the next best method this year.
Now recall what I said earlier about the rough meaning of different GDT_TS regimes and reexamine the plot. The improvement is nothing short of staggering and it is across the board. We see structures with a GDT_TS of 20, i.e., nonsense predicted by the next best method, getting to a GDT_TS of almost 90, i.e., with all the details!!! And then we see really good structures (mid 80s) predicted by the next best method going well north of 90 and 95 in some cases! Above 95 is within experimental accuracy.
It’s worth noting here that historically speaking it was very rare for one method to dominate others so thoroughly, certainly in recent memory. All the top groups, in particular the Baker and Zhang groups, are often running neck and neck. The only real exception to this was the last CASP, when the first AlphaFold did best for 1/3 of targets. This time, AF2 did best for 88 out of 97 targets!
Below is the comparison between the 2nd and 3rd best methods to illustrate this point.
This addresses some of the concerns that I’ve seen on social media from people unfamiliar with CASP, e.g., that there may be an overfitting issue; for starters, CASP organizers go to extraordinary lengths to get really difficult protein targets, ones that are quite different from known structures. I think it’s fair to say that the difficulty of the CASP free modeling (FM) category is harder than most structures deposited in the PDB and so in terms of real-world conditions, CASP proteins are actually harder than usual. But seeing the deltas above further allays these concerns, given how badly (relatively speaking) everyone else did with respect to AF2. I admit this was a concern of my own, that perhaps this year was an “easy” year, as there is always some variability in target difficulty from year to year. Thankfully the CASP14 organizers quantified the difficulty of this year’s targets and found them to be harder than those of the few previous CASPs, so this was a hard year!
I was also concerned that the impressive median numbers may be hiding some poor predictions at the bottom of the distribution. I knew about the median GDT_TS of 92.4 early on but it wasn’t until the weekend before CASP14 that I got access to the full distribution. It turned out that just a handful of structures, five to be exact, had a GDT_TS below 70 (out of 93 predictions made by AF2). This was remarkable: less than 10% of structures can be considered to not have the details right. Furthermore when one delves into these five, two turn out to be NMR structures and three are part of oligomeric complexes. NMR structures can be floppy reflecting the fact that these proteins don’t have well defined structures. As for oligomeric complexes, AF2 only predicts the structures of individual protein chains and so cannot be expected to reflect their oligomeric state.
I hope this communicates how thoroughly shocking AF2’s accuracy is. When looking at RMSD, a metric more commonly used by the broader biology community, AF2 achieves for Cα atoms an accuracy of <1Å 25% of the time, <1.6Å 50% of the time, and <2.5Å 75% of the time. When considering all atoms including those of side chains, the numbers are <1.5Å 25% of the time, <2.1Å 50% of the time, and <3Å 75% of the time. All but 7 of its predictions (out of 93, so 7.5%) are less than 5Å over all side chain atoms, including the cases I mentioned previously. I repeat, it is <5Å 92.5% of the time over all side chain atoms. And it is worth noting here that the model has residue-level error metrics, and they appear to be robust enough to have alerted the AF2 team when they did poorly on a SARS-CoV2 protein, so it may be the case that AF2 can warn users in the remaining 7.5% of cases but we don’t know that for sure yet.
On Twitter some quoted my CASP13 blog post to assert that I had predicted things correctly two years ago—I most certainly had not! In 2018 I expected us to get to a median of 4Å Cα RMSD by 2022, which if you look at where the next best group is, would be about right. Instead we got 1.6Å in 2020. This I did not expect to see until the decade’s end and I might have even said would not be achievable without physics-based approaches like molecular dynamics (MD).
A Solution?
Given all the above, does AF2 constitute a “solution” of one form or another? This will invariably be an exercise in semantics. Part of me wishes that DeepMind never used the term, that the CASP14 organizers didn’t, and that I didn’t, because all it has done is raise temperatures on all sides without advancing the discussion one bit. I used it because I thought it was justified, and still do, and so I will spend a bit of time here unpacking it but really there are better things to do with one’s time.
First, protein folding has often been used, especially in the lay media, as an umbrella term for different problems. I list a few that qualify:
Prediction of the structure of a single protein domain from sequence
Prediction of the structure of a single protein, possibly comprised of multiple domains, from sequence
Prediction of the structure of a multimeric complex
Prediction of the major conformations of a protein
Prediction of the dynamic folding pathway(s) of a protein
For each of the above, there is furthermore the question of whether a prediction is “pure”, i.e., made from only a single sequence, or whether additional information is used, most commonly homologous protein sequences but possibly homologous structures and even other forms of non-sequence-based experimental data. In addition, there is the question of whether a solution constitutes solving the bulk of the problem or all imaginable instantiations of it. For example, proteins can have metals and other co-factors that alter their structure. Does a solution need to address all of them? What about unnatural amino acids? Entirely de novo proteins that may fold to structures unseen in nature? Finally there’s the question of accuracy—how good is good enough? Less than 3Å? Less than 1Å? Less than 0.5Å? It gets complicated.
Here’s what I think AF2 can do: reliably (>90% of the time) predict to reasonable accuracy (<3-4Å) the lowest energy structure of vanilla (no co-factors, no obligate oligomerization) single protein chains using a list of homologous protein sequences, i.e., some version of the second bullet point above. It seems to deal with multi-domain proteins just fine but it hasn’t been thoroughly tested in this regard; this was surprising to me. Beyond that it can’t yet handle any of the corner cases and it’s not working from single sequences.
Does this constitute a solution of the static protein structure prediction problem? I think so but there are all these wrinkles. Honest, thoughtful people can disagree here and it comes down to one’s definition of what the word “solution” really means. Let me explain why I consider this a solution.
While the current list of caveats is a long one, making it seem that AF2 has a way to go before tackling all corner cases and elaborations, it is my expectation that this is not the case because the core intellectual problem has been solved. I believe that everything on the preceding bullet list, excepting the very last item (and possibly the second-to-last) is now an engineering rather than a scientific problem. That doesn’t mean it’s any less important or any less hard. I consider myself an engineer as much as I am a scientist, maybe even more so. But there is an important distinction between scientific and engineering problems that is pertinent to the discussion here: engineering problems can be exceedingly difficult, and require the marshaling of inordinate resources, but competent domain experts know the pieces that need to fall into place to solve them. Whether or not a problem can be solved is usually not a question in engineering tasks. It’s a question whether it can be done given the resources available. In scientific problems on the other hand, we don’t know how to go from A to Z. It may turn out that, with a little bit of careful thought (as appears to be the case for AF2), one discovers the solution and it’s easy to go from A to Z, easier than many engineering problems. But prior to that discovery, the path is unknown, and the problem may take a year, a decade, or a century. This is where we were pre-AF2, and why I made a prediction in terms of how fast we will progress that turned out to be wrong. I thought I had a pretty good handle on the problem but I could not estimate it correctly. In some ways it turned out that protein structure prediction is easier than we anticipated. But this is not the point. The point is that before AF2 we didn’t know, and now we do know, that a solution is possible.
My claim is that given this knowledge all the corner cases have become engineering problems. Some may prove to be harder, in terms of required effort as measured by human hours, than solving the core protein structure prediction problem. For example dealing with unnatural amino acids will probably be a long slog. In all likelihood we will never solve every corner case. In my view requiring that we solve every corner case to declare something a “solution”, especially something as difficult as protein structure prediction and that is not mathematics, means we have decided to never use that word for the problem. That’s a reasonable choice and people can make it. For me, the word still has relevance in the specific way I describe. It means that the bulk of the scientific problem is solved; what’s left now is execution. This may be harder than all that’s come before, but if we’re motivated enough, in terms of building the models and collecting the data, then we can solve those other problems. Protein structure prediction is at the 90% of the problem but 10% of the effort stage.
One item on the above bullet list does not fall into this category and ironically it is the one that gave this field its name: the actual dynamic process by which proteins fold. This is a completely different problem and I am almost tempted to say that AF2 has no bearing on it, but that would be far too strong I think—AF2, directly or indirectly, may well contribute to solving the protein folding problem. But that problem remains firmly in the realm of science. It may get solved in 5 years, or 10, or 100; we don’t yet know.
The Method
Now we finally get to something interesting: how AF2 actually works! Alas, I will be able to say a lot less than I had hoped for, and here I have to do something which I very much dislike to do but feel that I must—call out DeepMind for falling short of the standards of academic communication. What was presented on AF2 at CASP14 barely resembled a methods talk. It was exceedingly high-level, heavy on ideas and insinuations but almost entirely devoid of detail. This is a shame and contrasts markedly with DeepMind’s participation in CASP13, when they gave two talks that provided sufficient details for many groups to reproduce their results right away and participated in a poster session where they freely answered questions and built rapport with the community. While at CASP13 I and many others were surprised by DM’s entry and impressed by their results, we all walked away feeling like there’s a great new group of colleagues in the community. I’m afraid this time they left a different impression and I’m not sure it was at all necessary. DeepMind is in an exceedingly dominant position here—they will invariably get the cover of Nature or Science and may one day nab their first Nobel prize for AF2. Withholding details stands to poison the well of goodwill in the community. I hope their paper corrects it, and I furthermore hope they preprint their results to accelerate dissemination of their work.
Alright, enough with the rant! So what did they do? Insofar as I can tell, there were four major pieces to their scheme, which I will list in my perceived order of their importance. But I could be very wrong, on the order and on the details, as they really told us very little.
Out with the Potts models, in with raw MSAs
The current standard is to build a multiple sequence alignment (MSA) of homologous protein sequences then extract summary statistics out of this alignment, roughly speaking how strongly co-evolving every residue is with respect to every other residue. This summarized information is then fed into a neural network to predict a “distogram”, a matrix of the probabilities of pairwise distances between all Cβ atoms (sometimes other quantities are predicted but I’m simplifying). This was the approach of the first AlphaFold and multiple methods since then.
The new AF2 no longer summarizes the MSA. Instead, it keeps all raw sequences and iteratively “attends” to them. At step n, AF2 decides which sequences are worth looking at and which can be safely ignored and based on this predicts a distogram. At step n+1, AF2 uses the distogram to decide which sequences to attend to next, and based on them predict a new distogram. It does this multiple times. How many it wasn’t clear, but if one were to squint at DeepMind’s slides it appears to involve a few hundred iterations. In this way AF2 start outs building the local structure within individual protein domains before branching out to more global features, for example the relative orientation of two domains within a protein.
This approach is novel and has a number of potential advantages (simpler precedents do exist in the literature). First, AF2 can leverage deeper (as in having more sequences) MSAs for individual protein domains and shallower ones for whole proteins. It is quite common for individual domains to have a lot more sequences available and this can be used by the attention mechanism to resolve intra-domain details. Once done, AF2 can then use the relatively shallower full-length MSA to resolve the inter-domain details. I’m not proposing that this pre-engineered; AF2 likely learns how to do this on its own.
There are other, perhaps more significant advantages to this approach. MSAs can often be noisy, containing sequences that are not evolutionary related. By allowing the model to choose what to include at every step, AF2 can learn to filter on its own. Second, the summary statistics I previously described are all pairwise, extracting only how two residues co-evolve at a time. By accessing the full MSA, AF2 may be able to extract higher-order correlations. Third, some proteins, depending on the availability of data and their evolutionary age, can have very shallow MSAs. This has traditionally made it difficult for MSA-based methods to predict their structures (and what set me off in the first place to predict structures from individual sequences), including the first AlphaFold. This time around, AF2’s performance appears almost entirely decoupled from MSA depth. At least, it appears to be quite robust to proteins with very shallow MSAs. This may be due to the iterative attention scheme, as it is always operating on a self-chosen weighted mixture of individual proteins. When confronted with shallower MSAs it can learn to make do with what it has. Finally, the iterative approach is a good idea in general. We know from many machine learning tasks, especially in computer vision, that better results are achieved when a model is able to inspect its own output to generate refined outputs in response.
End-to-end differentiability
The second big change that DeepMind made is to reformulate the entire pipeline, from raw MSA to final predicted structure, to be end-to-end differentiable. On this point I feel justified in taking some credit for having developed the first end-to-end model for protein structure prediction (RGN). Mine was developed contemporaneously with another model (NEMO) by John Ingraham and colleagues (my preprint came out a few months before theirs but their final version was published a few months before mine). Both of our models did not perform competitively with MSA-based approaches as we relied on either single sequences or more limited forms of evolutionary information. That may have been a strategic mistake on our respective parts, but either way the details of AF2 are certainly very different.
End-to-end differentiability allows for model parameters to be tuned jointly, from beginning to end, to optimize for the final 3D structure instead of proximal quantities like inter-atomic distances. Second, and perhaps more importantly, it acts as a self-consistency constraint. Approaches that are not end-to-end generate outputs that can be inherently contradictory. For example, if the distances between all atoms are predicted simultaneously, they may not be embeddable in three-dimensional space, essentially giving nonsense. This is traditionally resolved by feeding the outputs through an optimization procedure, sometimes a physics-based one, which is what the first AlphaFold and all other methods did except for RGN and NEMO. With an end-to-end approach, the model, one way or another, must figure out how to be self-consistent. In the RGN case this was by construction because it reasoned in internal coordinates and iteratively built the protein structure one atom at a time so that it never made inconsistent predictions. AF2 doesn’t appear to do this, and it is unclear how it achieves self-consistency, as the fundamental object it operates on during the iterative attention portion is the distogram. At some point the distogram gets converted into 3D coordinates to be fed into the structure module (described next) but how that leap is made is left to our imagination. There are certainly known ways to do it and the answer may be vanilla but as of now we don’t know.
Iterative refinement using SE(3)-equivariant transformers
At some point, either after a fixed or learned number of iterations in distogram-space, AF2 generates what is likely a 3D point cloud that is then fed into an SE(3)–equivarianttransformer. I will explain what this means momentarily but for now suffice it to say that they use machinery that operates directly on atoms in 3D space. This is important because it captures higher-order coordination between atoms that cannot be captured by distograms, which are always about two atoms at a time, or at least two contiguous stretches of atoms. In 3D, multiple atoms from distant protein regions can all come together, and they are presented to the model to operate on and refine. This is an approach that we have also been working on for the last two years and so it is a bit painful to get scooped here. The computational requirements for equivariant neural networks can be substantial which slowed our progress.
Interestingly, the number of iterations that AF2 performs in 3D space seem to be on the order of 10. I say this is interesting because it is the geometric mean of what the RGN and NEMO models do. To elaborate: the original RGN did not iterate whatsoever, predicting a single structure in one shot. This made it very fast (milliseconds) but prevented it from performing any sort of refinement. NEMO went to the other extreme in a way, performing around 200 iterations using Langevin dynamics. It could slowly fold the structure but may have taken the physics too seriously by trying to emulate an energy landscape that can be traced down to its minimum. AF2 seems to do something in-between, both in the literal sense of taking 10 steps but also in formulating the refinement not as a physics-inspired process but as an iterative neural refinement process that “fixes” bad structures in likely non-physical ways (the fixing is non-physical, not the final structures). We don’t actually know the details and so I may be projecting here based on our own work but this is my best current guess of what AF2 is doing.
As for SE(3)-equivariant networks: proteins are molecules that exist in 3D space (in solution) without a preferred orientation or location (as individual abstract molecules). Much of the neural network machinery that’s been developed for images is (locally) translationally-invariant, i.e. does not care about location, but is not rotationally-invariant. The last few years have seen a mini-explosion of neural primitives that respect rotational invariance (equivariance just means one keeps track of the rotation/translation instead of ignoring it). It was started by a seminal paper by Nathaniel Thomas, Tess Smidt, and others and has now evolved into a vibrant subfield exemplified by the works of Taco Cohen, Max Welling, and Risi Kondor, among others. The unifying theme is the fact that standard convolutions have a group-theoretic structure, traditionally Z2 or Z3, that can be generalized to other groups including the Lie groups that respect the type of rotational symmetry desired here. Very recently a paper by Fabian Fuchs et al. ported this idea to transformers, which don’t rely on the locality assumptions made by convolutions. Given the timing of the paper, it is unlikely that the same exact approach was used by AF2 (the authors were not part of the AF2 team) but the idea is probably similar.
Structural templates
The final new piece in AF2 harkens back to the olden days of protein structure prediction, when nothing worked and the only option was to crib off homologous structures. It’s a bit of a “cheat” but can be effective. AF2 incorporates this technique by taking structural homologs directly as inputs along with the MSA. Interestingly, the second best performing team this year, from the Baker lab, does something similar. It is unclear how much this trick is contributing to AF2’s performance as it is able to do well even on proteins with no structural homologs (in the global sense, i.e., that cover most of the protein—essentially all protein structure prediction nowadays is some version of local structural homology as we have likely experimentally saturated the space of all possible protein fragments on a coarse-grained level.)
Compute
There is one other aspect of AF2 worth commenting on that doesn’t have to do with the approach per se but is interesting in its own right and is something of a mystery to me: the compute resources used. For training, AF2 consumed something like 128 TPUs for several weeks. That’s a lot of compute by academic standards but is not surprising for this type of problem. What is surprising however is the amount of compute needed for inference, i.e., making new predictions. According to Demis Hassabis, depending on the protein, they used between 5 to 40 GPUs for hours to days. Although they phrased this as “moderate” it is anything but for inference purposes. It is in fact an insane amount given that they are not doing any MD and has me perplexed. Nothing in their architecture as I understand it could warrant this much compute, unless they’re initializing from multiple random seeds for each prediction. The most computationally intensive part is likely the iterative MSA/distogram attention ping-pong, but even if that is run for hundreds or thousands of iterations, the inference compute seems too much. MSAs can be very large, that is true, but I doubt that they’re using them in their entirety as that seems overkill. At any rate I wonder if there is something to be learned from this mystery or if I am missing something obvious.
Impact on …
Alright so AF2 is amazing and all and maybe/maybe not solved protein structure prediction. What does this mean for …
Protein structure prediction?
The core field has been blown to pieces; there’s just no sugar-coating it. I can say this because it’s (one of) my own field(s). There are some intellectually interesting exercises left, for example predicting structure from a single sequence without structural templates or evolutionary information, and there are important engineering problems including addressing all the corner cases that AF2 still can’t. These are important and scientifically worthwhile but will be of limited interest beyond the core community of structure predictors. The “pure” problem of going from a single sequence to structure is the problem that’s been closest to my heart for over a decade, so it’s painful to say it, but it is the truth. It is similar to how the first mathematical proof of a result garners the most interest and accolades, even if subsequent complementary proofs are interesting in their own right. Everyone in the field now will be faced with tinkering around the edges, finding second proofs, or leaving to greener pastures. This was captured poignantly by a panelist at the very last session of the conference who remarked that CASP14 feels a bit like when one’s child leaves home for the very first time. It is good, in the sense that they have now matured, as has the field, and are ready to tackle bigger and better problems. But there is a bittersweetness to the experience that cannot be ignored.
To be clear I am referring only to the core protein structure prediction problem and not any of the proximal problems, including the ones on the bullet list in “A Solution?”. But, it’s going to be very risky for an academic group to swim in these waters. I won’t say more here as this is the topic of the last section of the blog post.
Experimental structure biology?
After the field of protein structure prediction itself, the fields that will most obviously be impacted by AF2 are those comprising the experimental determination of protein structure in various forms. In the most immediate term I suspect that even X-ray crystallography will actually benefit because of AF2’s value in molecular replacement, already discussed and demonstrated at CASP14. But beyond the short-term (>3-5 years) I expect AF-like models will begin to undercut crystallography. There were comments on Twitter to the effect of “call me when this hits 0.2Å” but that’s missing the point in my opinion. There are many, many, many applications in biology that do not require sub-angstrom accuracy and that will benefit a great deal from having structures even at 3Å or 4Å accuracy (not to mention, many crystallographic structures are not at 0.2Å resolution—but it’s important to note that we’re comparing apples to oranges here as prediction RMSD and crystallographic structure resolution are two different things.) Many of these applications formerly relied on crystallography because there were not many alternatives. Now that there will be soon, demand for crystallography will invariably fall. Of course it’s unlikely to ever get completely obsoleted, as few things ever do; some people still listen to the radio and ride horses, but it’s fair to say that this is not where the action is. I say this with empathy as someone who is contending with these questions myself—at least for most experimentalists, they can continue to do tomorrow what they were doing yesterday and this will remain the case for the next several years. For structure predictors they now have to instantly pivot or face obsolescence.
To be sure there will initially be justified reticence by the broader biology community to accept AF2 predictions as “truth”, but as we get to the point where the umpteenth crystal structure is produced and is in agreement within an angstrom or two of an AF2 prediction except for one tiny loop, attitudes will change. Maybe it will take more than a few years but the writing is on the wall. The applications that remain will require very high resolution, including most prominently drug discovery, and those will take longer to get affected. For some (very unscientific) support of this assessment I ran a poll on Twitter asking how excited biologists were by AF2’s specifications. Around 80% said they were pretty or very excited, while ~10% felt “meh” about the results. The latter is important as it suggests that for at least some subset of applications, AF2 remains underwhelming and this is a safe area for experimental methods for now. On the other hand, the 80% who were excited suggest that many existing needs may be met by AF2.
On the subject of crystallography, there is another point to note here—the cytoplasm is not a crystal! Most crystallized protein structures are probably not particularly good representatives of physiologic state! We’ve all been engaged in a sort of make-belief exercise that crystal structures give us the “truth”, and they do in a sense, about proteins coerced into forming crystalline material. But insofar as we’re interested in biological function, if predicted structures are well within the lowest energy basin of a protein’s energy landscape, I would argue it’s unclear whether crystal structures or predicted ones will be more informative. I mean that—it’s a question to be settled in the years ahead, especially as AF2-like predictions are coupled with MD methods to get more complete pictures of low energy ensembles. Hence I find some of the hand-wringing about crystallography being the ultimate arbiter of truth a bit off-the-mark given its own limitations. Of course the one big wrinkle in all this, at least for the time being, is that AF2 itself is trained on crystallographic structures in the PDB. And so it’s more accurate to think of it as predicting crystal structures as opposed to predicting the lowest energy state of proteins. This is an important caveat and will only be addressed when other experimental techniques and physics-based computational methods are systematically integrated. This is an exciting frontier and one that may actually be a good space for academics to play in, as it won’t leverage DeepMind’s ML expertise as much as AF2 has.
Speaking of other experimental techniques, next up is single particle CryoEM. The outlook for CryoEM is in my opinion better in the short and medium terms, as CryoEM is increasingly focused on quaternary complexes and molecular machines. If anything AF2 will help CryoEM because it’s the details of the individual monomers that CryoEM struggles with and AF2 excels at. So a win-win. Still, DeepMind has made it clear that complexes are their next big target, and I do think that most of the intellectual heavy lifting is done, hence the “solved” part. Going from monomers → complexes will, I predict, be easier than going from pre-AF2 → monomers. The only real question is how much of a negative impact will the relative paucity of experimentally-determined structures of quaternary complexes make on AF2-like models. This could turn out to be a serious issue but I wouldn’t bet on it, and at any rate structures of quaternary complexes are being generated at an accelerating rate.
The one area that I do think is truly the future of experimental structural biology, and which will remain safe and wholly complementary to AF2, is in situ structural biology. Getting the cellular context of structures is not something that DeepMind can meaningfully tackle anytime soon, but helping identify structures in their cellular context is definitely something that AF2 can help with. If anything AF2 may accelerate the breakneck pace of progress in CryoET and usher in the era of structural cell biology faster than even its proponents are expecting. I know a few people at EMBL who will be happy to hear this!
Biology as a whole?
This is the section I have been most looking forward to write for it is here that we can begin to imagine what can be.
While I love structural biology, and love staring at proteins, my interest in the field has always had a practical bent: structure not for its own sake but in service to biology. For this vision to become reality we need data, structural data, which has always been very hard to come by. AF2 is profoundly transformative because it may do for structure what DNA sequencing did for genomics; make it possible. Every question in biology, from the molecular to the cellular to the organismal to the evolutionary, can now be posed and framed in terms of structural hypotheses. We’ve done this with sequence for at least a couple of decades and it has come to define every facet of biological sciences. Now we get to do it over all again with structure. And while the structure → function dogma never fully rung true with me, it’s certainly the case that having structure > sequence for determining function.
So what does all this mean on a practical level? Truth be told, I don’t really know, and I suspect most of us don’t. We have yet to fully grok the consequences. I have been thinking about this question ever since CASP13 but have yet to truly internalize it. When televisions first appeared networks simply emulated radio broadcasting by having talking heads fill the screen. We will do the same for a while because we don’t understand what structure at scale means. But in due time we will. I am biased of course, but I believe this is the most important question facing basic biology in a post-AF2 era.
Still, we can speculate. First is the question of function derived from structure. There have been numerous efforts to predict protein function and do so in nuanced ways that reflect their multifunctional reality. Thus far these efforts have largely relied on sequence—now all can be redone using structure. For some protein classes, particularly enzymes, this may substantially improve accuracy, especially if the structures are good enough to resolve catalytic sites. Even if they are not, coarse structures will help relate proteins in the “twilight zone”, i.e., ones far from anything we’ve functionally characterized, to ones we do know something about. Especially in prokaryotic biology, where vast swaths of bacterial proteomes are still entirely uncharacterized, this alone may transform our ability to understand and one day engineer them.
It won’t happen overnight. None of what I’m saying here will. It will take years and maybe decades, but now that protein structure prediction has become an engineering exercise, we know that many of these ideas can be realized.
What else? Variant prediction. Interpreting what mutations do to proteins is both hard and important for human disease. So far we’ve been restricted almost exclusively to statistical approaches. We can’t predict what a mutation does to a protein on a molecular level, but we can discern whether a mutation is deleterious from looking at the genetics of healthy and sick populations. Because many diseases arise from interactions between and mutations in multiple genes, piecing their puzzles together requires extraordinary statistical power save for the simplest “Mendelian” diseases. Having structures for most proteins won’t, again, solve the problem overnight, but it will provide us with more powerful tools. This is especially true if we can predict how mutations alter not only individual proteins but their interactions as well. One major caveat here is how well AF2 captures the impact of small changes in sequence on structure, especially because it is an MSA-based method. A month ago I would have predicted not very well. Today I am not sure, but it will be a key test of its capabilities. Even if AF2 turns out to underperform on this problem however, I believe we now have the tools to build future versions that can address the problem.
What else? Possibly protein design. Like with variant prediction, we don’t have direct evidence that AF2 can do this, although Demis Hassabis did mention it as one of their goals. Two years ago I would have been somewhat pessimistic, both because AF2 is trained mostly on natural proteins and because it requires MSAs as input, which are not available for proteins that have undergone neither natural nor synthetic evolution. Now I am no longer so sure, primarily due to a series of three groundbreaking papers from the BakerandOvchinnikov labs that have utilized trRosetta, a system very similar to the first AlphaFold, to successfully design proteins. This bodes well for AF2. Nonetheless, I do suspect that AF2 has learned something like the “natural manifold” of protein structure space and may struggle with structures that look nothing like natural proteins, which to be fair no de novo protein design tool can yet do.
What else? Comparative “structuromics” (?!) Take your protein of interest and all the organisms in which it resides and examine how it has changed across evolutionary time. And all the proteins it interacts with and how they changed across evolutionary time. Couple that with functional characteristics of the protein itself, of its cellular context, and of its organism. Can we understand how changes in e.g. the structure of metabolic enzymes altered the metabolism of their organisms? What about cytoskeletal proteins and the morphology of their organisms? Signaling proteins and the information processing machinery of their organisms? And on and on.
What else? Synthetic biology,a field with tremendous potential hamstrung by its focus on engineering transcriptional circuits. Engineering DNA-based circuitry has been a natural choice because DNA is a lot more predictable than proteins, enabling the mixing and matching of promoters and DNA binding motifs with relative ease. But a cell can only do so much with transcriptional regulation, which is (likely) why it evolved a rich repertoire of information processing machinery in the form of protein-based signal transduction pathways as well as the structural machinery that gives the cell its form, motility, and function. Little of this machinery has been engineerable. Pioneering work by people like Wendall Lim mixed and matched modular protein domains to rewire signaling pathways but such systems remain brittle and difficult to engineer, requiring trial and error that is not dissimilar from how we used to build bridges before the advent of civil engineering. AF2 stands to change this, not just in terms of de novo protein design but also in engineering multi-domain proteins with flexible linkers and programmable logic. Like in other applications, it remains to be seen whether AF2’s performance broadly generalizes to the multi-domain context, but if it does, it will open up new opportunities complementary to existing protein design efforts. Once engineering protein-based circuits is feasible, it is not hard to imagine graduating to the next level in complexity.
Which brings me to what I think is the most exciting opportunity of all: the prospect of building a structural systems biology. In almost all forms of systems biology practiced today, from the careful and quantitative modeling of the dynamics of a small cohort of proteins to the quasi-qualitative systems-wide models that rely on highly simplified representations, structure rarely plays a role. This is unfortunate because structure is the common currency through which everything in biology gets integrated, both in terms of macromolecular chemistries, i.e., proteins, nucleic acids, lipids, etc, but also in terms of the cell’s functional domains, i.e., its information processing circuitry, its morphology, and its motility. A structural systems biology would take this seriously, deriving the rate constants of enzymatic and metabolic reactions, protein-protein binding affinities, and protein-DNA interactions all from structural models. We don’t yet know how much easier, if at all, it will be to predict these types of quantities from structure than from sequence—we need to put the dogma of “structure determines function” to the test. Even if the dogma were to fail in some instances, which it almost certainly will, partial success will open up new avenues.
Systems biology has hitherto been surprisingly non-spatial. ODEs over PDEs as it were. There are many reasons for this beyond the issue of structure, but as in situ structural biology becomes increasingly more powerful and is combined with predicted protein structures, new forms of simulation become possible that do take space seriously, both on the microscale of molecular machines as well as on the mesoscale of cellular components. These are long-term visions likely to take a decade or decades but the groundwork for them has been laid with AF2. Up till now structure only existed in small isolated pieces, for fragments of proteins and fragments of proteomes. AF2, when it becomes widely available, will change all this.
Drug discovery?
I will end this section with the question that gets asked most often about protein structure prediction—will it change drug discovery? Truthfully, in the short term, the answer is most likely no. But it’s complicated.
One important thing to note is that, of the entire drug development pipeline, the early discovery stage is just that, an early stage. Even if crystallography were to become fast and routine, it would still not fundamentally alter the dynamics of drug discovery as it is practiced today, as most of the cost is in the later stages of drug development beyond medicinal chemistry and well into biology and physiology. Reliable protein structure prediction doesn’t change that.
In my CASP13 post I took pharmaceuticals to task for not investing in protein structure prediction. This was not because it has immediate applications, certainly not back then. Instead, I thought that a problem of such fundamental biochemical importance ought to interest pharmaceuticals if for no other reason than to develop a robust basic research program that attracts the world’s best talent, especially the world’s best machine learning talent. This is arguably the real value proposition of DeepMind for Google (and MSR for Microsoft, FAIR for Facebook, etc.) Not immediate translation, but an intellectual core that feeds into other parts of the company. Some pharmaceuticals may be beginning to see the value of this but most of the exciting work remains in startups and companies backed by forward-thinking VCs like Flagship Pioneering and a16z.
Getting back to early-stage drug discovery, one part where AF2 can help is in determining structures of protein targets that can be modulated for therapeutic purposes. The challenge here, for the very immediate future, is that AF2 is trained to predict apo (unbound) protein structures while most medicinal chemistry applications require complexes of the protein bound to a small molecule. Second, sub-angstrom resolution is often necessary, which remains beyond what AF2 can achieve. A more fruitful direction for AF2 may lie in designing protein-based therapeutics, e.g., antibodies and peptides, where ultra-high resolution is less needed.
In the long run the true power of AF2 may come in providing a more robust platform for drug discovery, particularly within a systems pharmacology framework. We’re not there yet but we can imagine a future in which drugs are designed for their polypharmacology, i.e., to modulate multiple protein targets intentionally. This would very much be unlike conventional medicinal chemistry as practiced today where the emphasis is on minimizing off-targets and making highly selective small molecules. Drugs with designed polypharmacology may be able to modulate entire signaling pathways instead of acting on one protein at a time. There have been many fits and starts in this space and there is no reason to believe that a change is imminent, especially because the systems challenges of the equation remain formidable. Wide availability of structures may hasten progress however.
Why DeepMind?
I promised to write one piece of armchair sociology and so here it is. Why was it DeepMind, rather than an academic group, that built AF2?
First and foremost it has to do with the people who make up the AF2 team. One should not pretend that they are substitutable. Even within DeepMind, if it were a different set of people we would likely have had a different outcome. This may seem obvious but I repeatedly heard people treat the AF2 team as an amorphous blob. Let us not forget that the main reason they did so well is because of who they are, their talents, and their dedication. In this most important sense, it is not about DeepMind at all.
Resources also helped and this is not to be underestimated, but I would like to focus on organizational structure as I believe it is the key factor beyond the individual contributors themselves. DeepMind is organized very differently from academic groups. There are minimal administrative requirements, freeing up time to do research. This research is done by professionals working at the same job for years and who have achieved mastery of at least one discipline. Contrast this with academic labs where there is constant turnover of students and postdocs. This is as it should be, as their primary mission is the training of the next generation of scientists. Furthermore, at DeepMind everyone is rowing in the same direction. There is a reason that the AF2 abstract has 18 co-first authors and it is reflective of an incentive structure wholly foreign to academia. Research at universities is ultimately about individual effort and building a personal brand, irrespective of how collaborative one wants to be. This means the power of coordination that DeepMind can leverage is never available to academic groups. Taken together these factors result in a “fast and focused” research paradigm.
AF2’s success raises the question of what other problems exist that are ripe for a “fast and focused” attack. The will does exist on the part of funding agencies to dedicate significant resources to tackling so-called grand challenges. The Structural Genomics Initiative was one such effort and the structures it determined set the stage, in part, for DeepMind’s success today. But all these efforts tend to be distributed. Does it make sense to organize concerted efforts modeled on the DeepMind approach but focused on other pressing issues? I think so. One can imagine some problems in climate science falling in this category.
To be clear, the DeepMind approach is no silver bullet. The factors I mentioned above—experienced hands, high coordination, and focused research objectives—are great for answering questions but not for asking them, whereas in most of biology defining questions is the interesting part; protein structure prediction being one major counterexample. It would be short-sighted to turn the entire research enterprise into many mini DeepMinds.
There is another, more subtle drawback to the fast and focused model and that is its speed. Even for protein structure prediction, if DeepMind’s research had been carried out over a period of ten years instead of four, it is likely that their ideas, as well as other ideas they didn’t conceive of, would have slowly gestated and gotten published by multiple labs. Some of these ideas may or may not have ultimately contributed to the solution, but they would have formed an intellectual corpus that informs problems beyond protein structure prediction. The fast and focused model minimizes the percolation and exploration of ideas. Instead of a thousand flowers blooming, only one will, and it may prevent future bloomings by stripping them of perceived academic novelty. Worsening matters is that while DeepMind may have tried many approaches internally, we will only hear about a single distilled and beautified result.
None of this is DeepMind’s fault—it reflects the academic incentive structure, particularly in biology (and machine learning) that elevates bottom-line performance over the exploration of new ideas. This is what I mean by stripping them from perceived academic novelty. Once a solution is solved in any way, it becomes hard to justify solving it another way, especially from a publication standpoint.
To be sure I would far rather have DeepMind be in this space than not, and I would not trade AF2 for the thousand flowers I mentioned; better a bird in hand. But it does raise the question of whether it is possible to have one’s cake and eat it too. To have fast and focused efforts co-exist with the slow and steady progress of conventional research.
Academic Research in a New Age
Speaking of co-existence, what are the prospects for academic research on biomolecular machine learning in the post-AF2 era? I write this section with an eye toward helping prospective researchers, students, and postdocs chart their path through what has become intensely competitive territory. After CASP13 I described the AlphaFold group as a “world class research team … competitive with the very best existing teams.” It’s fair to say that the situation now looks markedly more lopsided: DeepMind has become so dominant it should make any prospective researcher take pause before entering the field.
To think through what DeepMind’s presence means moving forward let’s place ourselves in DeepMind’s shoes and try to reverse-engineer their logic. My hope is that in so doing we begin to map out a research perimeter that is not under the constant threat of being crushed in <2 years’ time.
First some observations. DeepMind cares about making a splash. All their major efforts ranging from Go to StarCraft 2 to AlphaFold have been coupled with massive media blitzes. They do carry out more conventional and less glamorous research, but the projects with the big resources tend to be splashy ones.
Second, and this is a point that Demis himself made, DeepMind likes well-defined problems with clear objectives and metrics. Science is almost never this way but protein structure prediction actually fit the bill perfectly. There is literally a leaderboard every two years. Scientific problems with this feature are likely to attract DeepMind’s attention.
Third, DeepMind does have a core competency and it is machine learning. By the late 2010s protein structure prediction had turned into an almost exclusively machine learning problem. It required some domain expertise, and the AF2 team composition reflected that a bit, but by and large the hard problems were machine learning ones. This suggests that problems in which machine learning is not the core nut to crack are also less likely to attract DeepMind’s attention.
Fourth, given point three, any problem that machine learning can tackle must have a lot of data, and representative data that cover a large swath of the problem space.
As DeepMind begins to reckon with what comes next after AF2 they are likely to focus first on problems that look a lot like protein structure prediction. Based on the above observations let’s consider some of these outstanding problems. The first and most obvious is predicting the structure of protein complexes. It is the “next step” after protein structure and is sufficiently proximal that CASP already has a category for it called Assembly. Data here is not as abundant as in protein structure prediction, in fact there is probably around an order magnitude less, but it is also the case that multi-domain proteins are somewhat informative of this problem as inter-domain packing is more similar to protein complex formation than protein folding. Based on this and DeepMind’s repeated assertions that they want to tackle the problem, it’s fair to say that this will be very competitive territory. My advice here, as will be elsewhere, is not to throw one’s hands in the air and exit the space altogether. Instead my suggestions are twofold. First, be aware of the leaderboard clock if it exists in your subfield and plan to publish off-cycle so as not to get crushed by DeepMind’s media machine. In some ways trRosetta is a good example of this, landing right in between CASP13 and 14. Second, work on developing general purpose machinery that can be applied to many problems. If quaternary complexes happen to be an open problem at the moment, by all means tackle them, but avoid constructing overly specialized toolkits that take a lot of time to develop and generalize poorly to proximal problems. I think this is good advice in general, but all the more so in a hypercompetitive landscape.
What comes next? Protein-ligand, protein-DNA, etc. Here we begin to see some cracks. Protein-DNA interactions probably check off enough boxes that DeepMind may well tackle them. Protein-small molecule interactions are very tempting of course, because of drug discovery applications, but the scientific problem is much hairier. One because organic molecules occupy a larger and more topologically complex chemical space than proteins. Two because the data is much worse, distributed across multiple silos, many behind corporate IP walls, and inherently much less randomly sampled and representative. It doesn’t make the problem impenetrable to DeepMind but it almost certainly means they can’t crack it in its full generality. They can aim at bits and pieces of it, which lowers the splashiness of any solution. I suspect that because of commercial implications they will make a serious push, but it will have less of an impact on academic research, resulting in work that gets published in specialized journals or that is entirely locked up from view.
What comes after that? Perhaps protein function prediction. What is protein function? That is in itself a good question, but asking questions is not the sort of challenge DeepMind wants, so let’s look for predefined notions of protein structure-function relationships. There are certainly some, for example the classification of enzymes into EC classes. This problem is unlikely to generate many CNS papers however. And even if “solved”, I would argue what would be really interesting is a finer-grained delineation of enzymatic categories and a tighter understanding of the relationship between structure and enzymatic function, including allostery and dynamics. Once again we’re venturing into territories where problems are poorly defined and the most intellectually stimulating work is about defining problems rather than solving them. As we exit the realm of protein complexes and protein-X interactions, we quickly run out of problems with clear objectives and large datasets and begin to encounter problems of ever shrinking scope and dataset size. This doesn’t mean that DeepMind will not pursue some of them. But as they do, given that none are grand challenges worthy of the whole team, they will begin to splinter their human resources to focus on disparate projects, ones that require not some insignificant degree of domain expertise. This will dilute their team and, in the long run, make DeepMind look increasingly more like regular academic groups, but with better funding, better resources, backing of world-class engineering, and some degree of cooperation between the various subgroups. That is certainly not a bad place to be! I expect that they will however become less scary, resembling more the version I described at CASP13 than the terrifying bulldozer they have become at CASP14.
There is one other problem worth discussing here and that is the dynamic process of protein folding itself. It’s fun to speculate on whether DeepMind will attempt to tackle it. On the one hand, it plays well to some of their expertise, certainly reinforcement learning, and is in some sense a grand challenge. On the other hand, there are multiple strikes against it. First, there is virtually no experimental data to benchmark against. Not much in the way of clear-cut metrics as well. Success is hard to define here, other than in terms of proxy applications, e.g., can one design a better drug, which is what outfits like DESRES and Nimbus have long made a bet on. Speaking of which, there is also serious competition in this space from well-established industrial labs. Second, it is a hard problem, a really hard problem, much more so than protein structure prediction. Because of lack of data as I mentioned but also because the fundamental object that one is trying to infer is inherently more complicated than the single lowest energy state of protein structure prediction. If any group is up to the task it is DeepMind but it would mean them expending substantially more resources on the problem than they already have, perhaps an order of magnitude more. Would they do that? Which brings me to my third point. The grand prize has already been won with AF2. DeepMind declared victory and said that the “protein folding” problem is solved. From a publicity or even Swedish committee standpoint, there is not much more to be gained, which speaks against allocating inordinate resources to double down on the problem. My guess is that the scientists will push for it, because it’s exciting, but management will push back, because it will seem pointless and overly expensive. Either way I believe it is safe for academics to operate in this space for the foreseeable future, as the problem really is quite hard and I don’t foresee DeepMind making AF2-like progress in <5 years. Needless to say however I’ve been proven wrong by them before!
I will close with a broad comment about one’s motivation for research and how that interacts with being in a competitive field. Some researchers do research because they like solving puzzles for their own sake and could care less about publishing in glossy journals or even having practical impact. If you are in this category I say carry on, as DeepMind’s presence will have little effect and if anything may be a source of inspiration and new ideas. Some researchers like the competition and the race, the thrill of trying to outdo someone else. If you are of this sort, DeepMind’s entry may be the biggest boon you could ever hope for, presenting a bigger fish than anyone else in the space. Keep that in mind though, as getting repeatedly crushed may not be much fun. Finally, some researchers do research to make the world a better place, enhance our understanding of natural phenomena and in so doing empower human interests, including human health. For people in this category competing with DeepMind does not make such sense because they are a very capable team and are likely to crack the problems they set their sights on. Working on the same problems seems redundant to me and one might well as dedicate one’s energy to other, understudied, problems.
Speaking for myself, I am a mixture of all three but lean most heavily on the second and third archetypes. In some ways I entered academic science to temper my competitive tendencies, as I know they can get out of hand. If I wanted to really compete, I would build a startup that takes on DeepMind, but this seems a little pointless given that DeepMind is already there. Hence for so long as I am doing science, I will look for areas that are interesting, impactful, and understudied, as all things being equal, I would rather make an impact that betters the world than one that is neutral. Different people are different of course and you will have to chart your own path given your interests and temperament. Good luck!
Acknowledgements
Thanks to Randy Read for pointing out my inconsistency when using the terms “accuracy” and “resolution”. I have now made my usage more consistent, reserving “accuracy” for prediction RMSD and “resolution” for crystallographic resolution.
AI, ML, and DataBiologySciencecomputational biologydeep learningprotein designprotein structurerepresentation learning
But it may well be semi-supervised. For some time now I have thought that building a latent representation of protein sequence space is a really good idea, both because we have far more sequences than any form of labelled data, and because, once built, such a representation can inform a broad range of downstream tasks. … Continue reading →
Show full content
But it may well be semi-supervised.
For some time now I have thought that building a latent representation of protein sequence space is a really good idea, both because we have far more sequences than any form of labelled data, and because, once built, such a representation can inform a broad range of downstream tasks. This is why I jumped at the opportunity last year when Surge Biswas, from the Church Lab, approached me about collaborating on exactly such a project. Last week we posted a preprint on bioRxiv describing this effort. It was led by Ethan Alley, Grigory Khimulya, and Surge. All I did was to enthusiastically cheer them on, and so the bulk of the credit goes to them and George Church for his mentorship.
Surge has already done a great job summarizing the paper in a Twitter thread and so I won’t spend much time explaining what we did. I will instead focus on what I found surprising during the course of this project and the implications of representation learning for protein informatics and, in the long run, the rest of protein science. I suspect that these types of representations will become foundational to how we understand proteins in the future, perhaps rivaling the impact they have had on natural language processing. I also think that, viewed as a conceptual playground, proteins will prove to be a more fertile ground for ideation in semi-supervised learning than other application domains. In short, I will make the case that if you’re an ML researcher working on semi-supervised learning, one of the best, if not the best domain to be working in is proteins. Conversely, if you’re a protein informaticist I would encourage you to pay close attention to semi-supervised learning!
But first, the paper itself. The basic idea of ‘UniRep’ is to train a neural network model to compress protein sequence space, in this instance by training a (multiplicative) LSTM to do next letter (amino acid) prediction, thereby mapping any arbitrary protein sequence to a fixed-length vector representation. The techniques are not necessarily new, although of course applying them to the protein context brings its own challenges. What is remarkable about this seemingly simple procedure is the power of the resulting representation. It does a surprisingly good job of predicting protein function across a diverse set of tasks, including ones structural in nature, like the induction of a single neuron that is able, with some degree of accuracy (ρ = 0.33) to distinguish between α helices and β strands (I suspect the network as a whole is far more performant at this task than the single neuron we’ve identified, but we didn’t push this aspect of the analysis as the problem is well tackled using specialized approaches.) What this suggests, which till now I find remarkable, is that by simply having to be compressive in its representation of protein sequence space, an RNN is compelled to build an internal detector of secondary structure; presumably because knowing about secondary structure makes it a more efficient compressor. It is not dissimilar from some of the recent results on sentiment analysis, and models like GPT-2 are even more breathtaking, but the power of the compressive principle to be so damn effective at inducing human-interpretable structure still manages to surprise me. Perhaps this is even more so when the modality is not obviously sensory, but something abstract (albeit structured) like protein sequences. I recall not long ago one of the grand poohbahs of machine learning saying that non-sensory sequences, specifically biological sequences, are unlikely to benefit from RNNs precisely for this reason. I think it’s safe to say now that they’ve been proven wrong.
Another aspect that I found particularly surprising is the power of the unsupervised signal. Across the set of tasks we tackled, UniRep was able to outperform the structurally-supervised RGN models I published last year. One would have thought that structural information, particularly for tasks like thermal stability, would be far more valuable than sequence information. And of course it’s likely the case that if one were to have as many structures as sequences, a structurally-supervised model would do better. But given the gap in available data, sequence trumps structure. This antagonism is superficial of course—the right thing to do would be to leverage the advantages of both, and it is an avenue we’re currently exploring.
A key feature of this type of representation learning is its induction of a global representation of protein sequence space. Much of the prior work in this space has focused on family-specific representations, for example VAE-based ones. While family-specific representations have proven to be very powerful, particularly for protein structure prediction and for predicting the effects of mutant variants, they have always left me feeling somewhat unsatisfied. I say this because from the perspective of data / sampling efficiency, they’re only able to exploit patterns observed within a single protein family. They fragment protein sequence space into local clusters and perform learning, unsupervised or otherwise, separately for each family. This sort of sample vs. model complexity tradeoff is emblematic of much of machine learning, and I’ve written about it before in the context of predicting SH2-mediated protein-protein interactions. On the one hand, if each protein family is learned separately, the complexity of the model is reduced. But the amount of data available is also reduced, fracturing the inherent universality of proteins into tiny phenomenological universes. On the other hand, a global model of protein sequence space is able to leverage all available data, but must learn something truly general, a much more demanding task that substantially increases model complexity. What is the right tradeoff—where is the sweet spot? My hunch has long been that a global model is likely to be more performant. And with UniRep, I believe we’re beginning to see this play out.
Beyond the question of performance, a global model of protein sequence space has the potential to be much more useful, by being more broadly applicable. The challenge we face in protein informatics, and protein science more broadly, is that our functional characterization of proteins is sparse and patchy. Certain protein families have had the benefit of deep functional characterization, both in terms of gross function (e.g. the plethora of mammalian signaling proteins) and in terms of detailed structural perturbation (e.g. as resultant from deep mutational scans). The GFP family of proteins studied in the UniRep paper is an example of one such family. This patchiness makes it difficult to say something truly useful about the vast majority of proteins (across Prokarya and Eukarya) because, to use an overextended metaphor, they exist in a dark region of sequence space. And the detailed characterization we have of a few families ends up being largely uninformative about this larger space, except in a vague conceptual way.
Global models of protein sequence space have the potential to change this because, if we can get them to work well, they can at a minimum help us see the connective tissue that underpins protein space, and thereby relate information about well-characterized protein families to poorly characterized ones. In essence, they provide a fancy version of k-nearest neighbors, by densely populating the empty space surrounding sparsely characterized proteins, enabling functional associations to be transferred from one protein to another much farther than previously possible. I believe something similar was occurring in my differentiable RGN models of protein structure, in particular when I moved from training models on individual protein sequences to PSSMs. What PSSMs provide is precisely this form of connective tissue, by forging links between seemingly far away proteins whose relationship would be undetectable by mere sequence similarity. By leveraging the evolutionary record, one is able to see how one protein relates to another, and I suspect this allowed RGNs to become much more performant with PSSMs. UniRep has the potential to do something similar without PSSMs.
Beyond this, global models of protein sequence hold the possibility of learning something truly general about proteins, that would move us beyond mere k-nearest neighbor matching to something more akin to a linguistics of proteins, decomposing protein sequences into their constituent functional and structural fragments. The fact that UniRep learned something about protein secondary structure is an indication that this is possible and already happening, without any supervision. This is important because unlike secondary structure, most of the principles of protein function (and perhaps structure) remain opaque to us, and so our ability to perform supervision will remain limited for the foreseeable future.
To drive home this point, below is a plot of the number of available protein sequences and structures over the past decade.
What should be obvious is that new protein sequences are being acquired at a much faster pace than structures, and the above holds true for pretty much any form of functional characterization of proteins. There is no assay that we can perform today that will close this exponentially increasing gap, certainly nothing on the horizon.
On the one hand this may seem depressing, but on the other hand, I believe it presents a unique opportunity for unsupervised and semi-supervised machine learning. I am aware of no other problem in which the gap between labelled and unlabeled data is this large and continually increasing. If unsupervised learning can be made to work somewhere, it ought to be here. I emphasize this point because I have followed unsupervised learning with some interest over the last few years, and found most applications to be somewhat uncompelling, in the sense that the increase in performance gained from e.g. unsupervised initialization of an RNN always seems to be marginal. In many applications this is further exacerbated by the fact that acquiring labelled data is not all that expensive, rendering the extra effort that goes into semi-supervised learning even less worthwhile. However, the gap between labelled and unlabelled data in most applications is on the scale of 1 to 2 orders of magnitude, at most. What we see in protein sequence vs. e.g. structure is a gap of 5 orders of magnitude. This suggests, again, that if unsupervised learning were to work anywhere, it ought to be on proteins.
Another advantage of proteins is the wealth of prior knowledge that can be exploited to construct sophisticated loss functions for unsupervised learning, instead of simple next letter prediction (approaches like BERT have gone beyond this, but the amount of unsupervised signals in NLP seem to be much more limited than proteins.) A recent paper from Bonnie Berger’s lab demonstrates this idea (on the whole this paper deserves a lot more attention than it’s received IMO—it’s really a very good piece of work. Thanks to Tami Lieberman for the pointer.) Instead of simply learning to predict a missing amino acid, they augment their system with an auxiliary loss that predicts the structural distance between two proteins, based on their SCOP classification. It’s a simple idea, but demonstrates the type of structured knowledge that exists for proteins (to be sure, in this case the loss signal requires actual protein structures, and so it’s not entirely unsupervised.)
These are early days in our understanding and formulation of protein sequence space. It is an incredibly rich object, with much known and even more that is unknown. For a long time the only way we could relate one protein to another was through explicit pairwise or multiple sequence alignments, which assumed a direct evolutionary relationship and induced a residue-to-residue correspondence. What we’re beginning to see now is the emergence of something more general, a way to think about proteins that is less concerned with their evolutionary relationships and more concerned with their fundamental functional and structural constituents. If we push this approach to its limits, we may end up with a theoretical science of proteins, one which spans not only the space of extant proteins but generalizes to heretofore unseen ones. The challenge to this vision is the (lack of) human interpretability of such representations. If and how this challenge may be overcome, and whether it’s worth trying at all, is a subject for another post.
Acknowledgments: Thanks to Surge Biswas, Grigory Khimulya, and Ethan Alley for reading and providing feedback on an earlier version of this post.
AI, ML, and DataBiologyConferencesScienceCASPCASP13deep learningDeepMindGoogleprotein foldingprotein structure
Update: An updated version of this blogpost was published as a (peer-reviewed) Letter to the Editor at Bioinformatics, sans the “sociology” commentary. I just came back from CASP13, the biennial assessment of protein structure prediction methods (I previously blogged about CASP10.) I participated in a panel on deep learning methods in protein structure prediction, as well … Continue reading →
Show full content
Update: An updated version of this blogpost was published as a (peer-reviewed) Letter to the Editor at Bioinformatics, sans the “sociology” commentary.
I just came back from CASP13, the biennial assessment of protein structure prediction methods (I previously blogged about CASP10.) I participated in a panel on deep learning methods in protein structure prediction, as well as a predictor (more on that later.) If you keep tabs on science news, you may have heard that DeepMind’s debut went rather well. So well in fact that not only did they take first place, but put a comfortable distance between them and the second place predictor (the Zhang group) in the free modeling (FM) category, which focuses on modeling novel protein folds. Is the news real or overhyped? What is AlphaFold’s key methodological advance, and does it represent a fundamentally new approach? Is DeepMind forthcoming in sharing the details? And what was the community’s reaction? I will summarize my thoughts on these questions and more below. At the end I will also briefly discuss how RGNs, my end-to-end differentiable model for structure prediction, did on CASP13.
“What just happened?” was a question put to me in exactly these words by at least one researcher at CASP, and a sentiment expressed by most academics I spoke with. As one myself, I shared it going in and throughout the meeting. In fact I went into CASP13 feeling melancholy (the raw results were out two days prior), although my mood lifted during the meeting due to the general excitement and quality of discussions, and as my tribal reflexes gave way to a cooler and more rational assessment of the value of scientific progress.
This will be a long post. I will start with the science: the significance of DeepMind’s result, their methodology, and how it relates to existing methods. Then I will discuss the sociology: how people reacted, why we did so, what this means for the academic discipline of protein structure prediction (and life science companies), and how I think we ought to move forward. After what I hope is an exposition of general interest, I will briefly discuss how RGNs performed at CASP13. Spoiler alert: not very well, partly because the value of co-evolutionary information increased substantially in this CASP relative to prior ones, and partly because I could not submit the original submissions unaltered owing to technical problems.
For the sake of making this post easier to navigate, below is a table of contents.
Update: Jinbo Xu kindly wrote a number of thoughtful points in the comments section below, particularly here and here. They are well worth reading.
Let me get the most important question out of the way: is AlphaFold’s advance really significant, or is it more of the same? I would characterize their advance as roughly two CASPs in one (really ~1.8x). Historically progress in CASP has ebbed and flowed, with a ten year period of almost absolute stagnation, finally broken by the advances seen at CASP11 and 12, which were substantial. What we’ve seen this year is roughly twice as much as the recent average rate of advance (measured in mean ΔGDT_TS from CASP10 to CASP12—GDT_TS is a measure of prediction accuracy ranging from 0 to 100, with 100 being perfect.) As I will explain later, there may actually be a good reason for this “two CASPs” effect, in terms of the underlying methodological breakdown. This can be seen not only in the CASP-over-CASP improvement, but also in terms of the size of the gap between AlphaFold and the second best performer, which is unusually large by CASP standards. Below is a plot that depicts this.
Curves show the best and second best predictors at each CASP, while the dashed line shows the expected improvement at CASP13 given the average rate of improvement from CASP10 to 12. Ranking is based on CASP assessor’s formula, and does not always coincide with highest mean GDT_TS (e.g. CASP10.) Error bars correspond to 95% confidence intervals.
Prior to CASP10, for roughly ten years, the curve was basically flat. CASP11 began to show life because of the introduction of co-evolutionary methods, but just barely because most FM targets had shallow multiple sequence alignments (MSAs), which are required for co-evolutionary methods. CASP12 was when the power of these methods finally got demonstrated, and CASP13, even when excluding AlphaFold, showed further progress due to the widespread adoption of deep learning in co-evolutionary methods. We see that the second best method (Zhang server) improved by almost exactly “one expected CASP”, reflective of the field-wide improvement, and AlphaFold added to this yet another “one CASP”’s worth of improvement. Note these “one CASP”s are very recent history dependent, really just the past few CASPs (10-12), and so please take them with a mountainful of salt. Note also that my method of using mean GDT_TS is problematic because the difficulty of FM prediction targets varies from one CASP to another, although they’ve been supposedly stable recently.
Taken together the above suggests substantial progress, more so than usual, and hence not only did AlphaFold “win” CASP13, but did so by an unusual margin. Great! Does this mean the problem is solved, or nearly so? The answer, right now, is no. We are not there yet. However, if the (AlphaFold-adjusted) trend in the above figure were to continue, then perhaps in two CASPs, i.e. four years, we’ll actually get to a point where the problem can be called solved, in terms of gross topology (mean GDT_TS ~ 85% or so). Of course, this presupposes that the trendline will continue, and we have no real reason to believe that it will, at least not without new conceptual breakthroughs. Keep in mind that unlike other areas of machine learning, new protein structures are not appearing at an increasing rate, and so waiting things out will not help.
The above graph is misleading in one way though because it is dependent on a specific metric, GDT_TS, which only measures gross topology. If we care about high resolution topology, which we certainly do for most practical applications, then a more appropriate metric is GDT_HA, and using it the picture looks a bit different:
Still a good trendline, but much further down from a “solution”.
Another caveat is that both of these metrics measure global goodness of fit, which is important in terms of the basic scientific problem, but is often not indicative of functional utility. Local accuracy, for example the coordination of atoms in an active site or the localized change of conformation due to a mutation, is what is often sought when answering broader biological questions. Global metrics hide local discrepancy by diluting it in the sea of generally good agreement between experimental and predicted structures.
Another way of thinking about this is asking whether the same headlines would have been generated had an academic group achieved the same increase in accuracy that DeepMind has. The answer is certainly not, and we have the CASP11 → CASP12 advance to confirm that, as it was about equal in absolute magnitude (and thus arguably harder coming from a lower starting point) but generated few if any headlines. DeepMind’s publicity machine certainly helped shine a bright light on their advance, which is frankly also good for the field as a whole.
None of this is to detract from the AlphaFold advance. It is an anomalous leap, on the order of a doubling of the usual rate of improvement, and portends very favorably for the future. But that future has yet to be realized. (I actually think people may have walked away a bit too optimistic from this CASP—a DeepMind joins the field for the first time only once, and the value added of their excellent engineering may not get repeatedly re-realized, but we’ll see.)
Prior work
Let me now switch gears and talk a bit about the landscape of protein structure methodology before AlphaFold’s arrival. I won’t talk much about RGNs here because in some ways they are much more unusual methodologically than AlphaFold is, and so the two are well separated in algorithm space.
AlphaFold is a co-evolution based method, building on the groundwork that has been laid in the past ~7 years by several academic groups. The basic idea is to extract so-called evolutionary couplings from protein MSAs by detecting residues that co-evolve, i.e. that have mutated over evolutionary timeframes in response to other mutations, thereby suggesting physical proximity in 3D space. The first batch of such approaches [2, 3, 5] predicted binary contact matrices from MSAs, i.e. whether two residues are “in contact” or not (typically defined as being within <8Å), and fed that information to simple geometric constraint satisfaction methods to fold the protein and return its 3D coordinates. (There is a pre-history to this field when overly simple statistical models were used to predict such contacts, dating back to the 90s, but I will not cover it as that generation of approaches was not successful and I am by no means trying to be comprehensive here.) This first generation of methods was a substantial breakthrough, and ushered in the new era of protein structure prediction that finally showed promise of working.
An important if expected development was the coupling of such binary contacts with more advanced folding pipelines such as Rosetta and I-Tasser, which resulted in better accuracy and were the state of the art until around the middle of 2016, or just before CASP12. The next major advance came from applying convolutional networks and deep architectures (residual networks) to integrate information globally across the entire matrix of raw couplings to turn them into more accurate contacts. Jinbo Xu’s group developed the first major (and experimentally serious) version of this approach, among others [1, 4, 6].
Which brings us to the present and AlphaFold. Only a few weeks before the CASP13 results became public, Xu published a preprint on bioRxiv that predicted inter-residue distances instead of binary contacts [7]. It used the same input (MSAs), and largely the same architecture as his CASP12 approach, but predicted probabilities over a discretized spatial range and then picked the highest probability one for feeding into CNS to fold the protein. Xu’s preprint showed significant promise on a subset of CASP13 targets, and the buzz among some of us was that Xu’s approach would win the competition. As it turns out, this seemingly simple change had a surprisingly profound impact, and forms one of the key ingredients of AlphaFold’s recipe.
AlphaFold
DeepMind has promised to publish a paper on AlphaFold, so the final and definitive description will have to wait for their paper, which I hope will be thorough. They have no plans to release the source code, and are unlikely to put up a public prediction server in the near term, although they appear open to considering it at some point. Having said that, they were generally forthcoming in discussing their method during CASP13, and appeared genuinely interested in sharing the approach with the community and ensuring that people can build on it. The sense I got was that they are in it for the science.
Just like Xu’s approach, AlphaFold uses a softmax over discretized spatial ranges as its output, predicting a probability distribution over distances (the details of the convolutional ResNet architecture are different, but it remains unclear how large a contribution these details made.) Unlike Xu’s approach, which tosses away these probabilities and only uses the most likely distance bin as input to CNS, AlphaFold uses the entire distribution as a (protein-specific) statistical potential function that is directly minimized to generate the protein fold. The key idea of AlphaFold’s approach is that a distribution over pairwise distances between residues corresponds to a potential that can be minimized after being turned into a continuous function. They initially experimented with more complex approaches, including fragment assembly using a generative variational autoencoder. Remarkably however, halfway through CASP13, they discovered that simple and direct minimization of their predicted energy function, using gradient descent (L-BFGS), is sufficient to yield a high accuracy fold. And so they essentially switched to this approach half way and it represents the essence of their final model.
This idea looks deceivingly simple but has rather profound implications. I think its simplicity may somewhat mask the difficulty with which it can be arrived to. More often than not in science, particularly physical sciences, a simple change in perspective can lead to surprising changes in outcomes. The paradigm of predict contacts → feed into complex folding algorithm was so entrenched in the field that it was difficult for most to see it as unnecessary (including for DeepMind’s team, which tried more conventional folding approaches before discovering that a simpler approach works just as well.) Much of the pushback I received toward my end-to-end differentiable approach was because it eschewed any sampling and directly folded the protein.
There are some important technical details. The potential is not used as is, but is normalized using a learned “reference state”, harking back to the old days of knowledge-based potentials like DFIRE and the Quasichemical potential (parenthetically, I wrote a couple of papers on the topic, developing what I think was the first ML-based potential for protein-DNA interactions.) This normalization evidently had a large impact. Furthermore, their potential is coupled with a more traditional physics-based potential and the combined energy function is what is actually minimized.
This idea of predicting a protein-specific energy potential brings AlphaFold’s approach into proximity to another approach, called NEMO, which is currently in open review at ICLR. While the submission is anonymous, it is fair to conclude that, given this talk, it’s been developed by John Ingraham, Adam Riesselman, Chris Sander, and Debora Marks. NEMO too generates a protein-specific energy potential that is then minimized to yield the final protein, but the similarities end there. AlphaFold generates the potential using a neural network, but once done, turns it over to a minimizer that operates independently and is not optimized jointly with the neural network. NEMO on the other hand turns the entire folding process into a differentiable Langevin dynamics simulator, and backpropagates from the final predicted structure through a few hundred steps of the simulator into the neural network variables. Additionally NEMO, like RGNs, only uses raw sequence information and PSSMs.
While the AlphaFold and NEMO approaches do harken back to knowledge-based potentials, they are different in a fundamental way. The knowledge-based potentials of yore (and current physics-based potentials like Rosetta) are universal, in the sense that they at least pretend to be applicable to any protein, and would yield the right result if enough sampling was done to find their minimum. Whether this is true or not in practice is a different matter. The protein-specific potentials of AlphaFold and NEMO are quite different beasts. They are entirely a consequence of the MSA (or sequence + PSSM) that they depend on. What they do is construct a potential surface, particularly in the case of AlphaFold, that is very smooth for the given protein family, and whose minimum closely matches that of the native protein (-family average) fold. It is fantastic (and surprising to some, but I would argue RGNs already showed it is possible by doing so implicitly in the RGN latent space) and extremely useful, in that it allows one to make accurate predictions given an MSA. But it is not an energy potential in the conventional sense.
I should say that this is my characterization and not DeepMind’s. In general I have fairly strong feelings about protein-specific energy potentials, and was planning on writing a more detailed blog post about the topic in connection with the NEMO paper, but have not gotten around to it yet (and unfortunately probably never will.)
Below is a table that summarizes my view of how all the approaches I have discussed so far relate. Adjacent columns in the table indicate methods that in some sense are most similar, but because this is a multi-dimensional space, the relationships are more complex than that. For example, Xu’s approach is similar to AlphaFold because of their prediction of distances, while NEMO is similar to AlphaFold because of their use of protein-specific energy potentials, while NEMO and RGNs are similar because they are end-to-end differentiable and don’t use MSA data, which puts them in a different category altogether. I should point out that NEMO did not participate in CASP13, and neither NEMO nor RGNs are broadly competitive with the other methods (particularly on templated-based modeling (TBM) for RGNs), at least in part because they are using a lot less information.
ZhangXuAlphaFoldNEMORGNInputsMSAMSAMSASequence or PSSMPSSMOutputs (pre-folding)ContactsDistancesDistributions over distancesCartesian coordinates (folding internal)Cartesian coordinates (folding internal)FoldingI-TasserCNSL-BFGSDifferentiable Langevin dynamicsImplicitEnergy functionExplicit, fixed, and universalNoneExplicit, learned, and MSA-specificExplicit, learned, and sequence- or PSSM-specificImplicit, learned, and PSSM-specificUses templatesYesNoNoNoNoEnd-to-end differentiableNoNoNoYesYes
The careful reader will note that one column in the above table covers the Zhang group method, which I have not talked about much. Zhang’s approach is interesting for several reasons. First, it came in second during CASP13, and when looking at the overall results (not just FM but also TBM), it is not that far behind AlphaFold’s method. Remarkably, Zhang’s approach does not use predicted distances, but relies on the old style binary contacts. This raises the question of where their improvement is coming from. There are several things going on. While Xu’s approach uses the more informative distances, its folding pipeline is rather simplistic. Zhang’s approach, while using the less informative binary contacts, folds via the sophisticated I-Tasser engine. Since the groups were working independently (and largely in secrecy and competitively), they did not add up their relative contributions. If it were not for AlphaFold, this combined “double” effect may not have been seen until CASP14, but AlphaFold effectively did both at once. Of course, the way AlphaFold achieves this is not via a better folding engine, as theirs is very simple too (L-BFGS). Rather, they get around the problem by building a better energy potential using distributional information. But the advantages of having such an energy potential may be compensated by using a stronger folding engine. I-Tasser also uses templates from the PDB which can substantially help its performance on TBM targets. And perhaps there is further gain to be had by combining AlphaFold’s approach with something like I-Tasser or Rosetta, but AlphaFold’s preliminary results seem to suggest that they’ve already squeezed out what can be had from a better folding engine.
This sheds some light on AlphaFold’s novelty (more on this next.) If it weren’t for AlphaFold, what the field may have moved towards is combining Xu’s approach with Zhang’s, which would have arguably been less elegant than AlphaFold. But this is highly speculative, and it is likely there’s a “half CASP” waiting to be squeezed out by leveraging these partially complementary approaches.
Fundamental scientific insight or superb engineering?
A question that arose over many conversations at CASP13 is whether AlphaFold represents a triumph of insightful science or superb engineering? Such questions can often be silly and divisive (with science somehow occupying a higher ethereal realm than engineering), but at the heart of the question is whether AlphaFold “only” won because it has a large and well-funded team with inexhaustible compute resources, and therefore the academic community has nothing to feel bad about and need not engage in uneasy introspection (you can tell I’m gearing up to shift to the sociology), or whether they have done good science that the academic community missed out on. Insofar as this question merits answering, my own take is that it’s a mixture of the two.
On the fundamental insight front, AlphaFold had a number of good ideas. First, don’t just predict contacts, but also distances, something that Xu does as well but all indications point to the two groups having independently developed the idea. Critically, AlphaFold takes this a step further by predicting a distribution over distances, and then uses that to construct a smooth potential that is minimizable. A second good idea is the use of a reference state, which debiases the predicted potential and demonstrates a solid understanding of knowledge-based potentials that reflects positively on the DeepMind team. The fact that these ideas are “simple”, in the sense that they are unsurprising does not detract from them in the least bit (personally I was actually surprised by the impact the reference potential made, but others appeared less surprised.) The best science is one in which simple ideas have profound consequences, and it very much appears to be the case here. DeepMind is of course also leveraging their deep (no pun intended) expertise in machine learning. For example, the distributional prediction idea seems somewhat similar in spirit to their paper from about a year ago on distributional RL. Whether that insight had any impact on AlphaFold I don’t know, but I think it’s fair to say that the confluence of strong expertise in ML and proteins helped to bring about these advances.
On the engineering front, it’s also clear that the apparently elegant solution we see now is a result of much trial and error, and that much more complex components involving fragment assembly and so on were tried and disposed of. The ability to explore model space rapidly depends heavily on both computational and human resources. So while the final ideas are simple and elegant, they are unlikely to have been discovered if the AlphaFold team wasn’t able to sweep through idea space as rapidly as they did.
If I were to pick, I think about half of the performance improvement we see in AlphaFold comes from the simple ideas above, and about half from the sophisticated engineering of the distance-predicting neural network. If this is true, then academic groups should be able to see substantial improvements in fairly short order.
The sociology
“What just happened?”
Now that the serious and respectable matters are out of the way, I can finally engage in some gossip. This part will be quite the rant. Like I alluded to in the very beginning of this post, there was, in many ways, a broad sense of existential angst felt by most academic researchers at CASP13, including myself. In a delicious twist of irony, we the people who have bet their careers on trying to obsolete crystallographers are now worried about getting obsoleted ourselves.
I think many of us went through the following phases: (i) fearing that the DeepMind team outsmarted us all by some brilliant fundamental insight, combined with virtuoso engineering; (ii) breathing a sigh of relief that the insights were not radically different from what most of the field was thinking; (iii) (slightly) belittling DeepMind’s contribution by noting its seeming incrementality and crediting their success to Alphabet’s resources.
Setting aside the validity of the above sentiments, the underlying concern behind them is whether protein structure prediction as an academic field has a future, or whether like many parts of machine learning, the best research will from here on out get done in industrial labs, with mere breadcrumbs left for academic groups. Truth be told, I don’t know the answer, and I think it’s possible that some version of this will come to pass. What is clear is that the protein structure field has a new, and formidable, research group. For academic scientists, especially the more junior among us, we will have to contend with whether it’s strategically sound for our careers to continue working on structure prediction. Despite the size of the Baker and Zhang groups for example, I never felt intimidated by them, because on the novelty front I always felt I was several steps ahead. But with DeepMind’s entry I will have to reconsider, and from conversations with others this appears to be a nearly universal concern. Just like in machine learning, for some of us it will make sense to go into industrial labs, while for others it will mean staying in academia but shifting to entirely new problems or structure-proximal problems that avoid head-on competition with DeepMind.
So that’s what just happened. What I’d like to turn my attention to now is what this episode says about academic science, particularly as it pertains to protein structure prediction, and the scientific health of pharmaceutical companies (prepare to be roasted!)
An indictment of academic science
I don’t think we would do ourselves a service by not recognizing that what just happened presents a serious indictment of academic science. There are dozens of academic groups, with researchers likely numbering in the (low) hundreds, working on protein structure prediction. We have been working on this problem for decades, with vast expertise built up on both sides of the Atlantic and Pacific, and not insignificant computational resources when measured collectively. For DeepMind’s group of ~10 researchers, with primarily (but certainly not exclusively) ML expertise, to so thoroughly route everyone surely demonstrates the structural inefficiency of academic science. This is not Go, which had a handful of researchers working on the problem, and which had no direct applications beyond the core problem itself. Protein folding is a central problem of biochemistry, with profound implications for the biological and chemical sciences. How can a problem of such vital importance be so badly neglected?
Part of the problem is the nature of academic research. Marc Kirschner recently framed this beautifully, and I will copy it here verbatim:
“I believe that science, at its most creative, is more akin to a hunter-gatherer society than it is to a highly regimented industrial activity, more like a play group than a corporation.” – Marc Kirschner
I wholeheartedly agree with this, and think it is a good thing. The problem occurs when we take this analogy to mean that each small unit of hunter gatherers must defend its turf at all costs, as if the acquisition of scientific knowledge is akin to the hording of food. Science is, in the final analysis, a collective enterprise, and we all gain the greatest benefit when we cooperate and share our knowledge. An element of competitiveness is unavoidable given the human nature of this activity, but it should not rise to the toxicity that currently characterizes much of academia.
More important, and this is where the protein structure field has a very serious problem, the sharing of information must occur with frequent regularity. Even if individual groups are secretive while carrying out their research, if the frequency of sharing is on the order of months, as is typically the case in machine learning, the field can still progress at a rapid pace. But in part due to the canonicalization of CASP, protein structure prediction effectively has a two-year clock cycle, where separate research groups guard their discoveries until after CASP results are announced. As I discussed earlier, it is clear that between the Xu and Zhang groups enough was known to develop a system that would have perhaps rivaled AlphaFold. But because of the siloed nature of the field, it only gets a “gradient update” once every two years. Academic groups are thus forced to independently rediscover the wheel over and over. In DeepMind’s case, even though the team was small in comparison to the total headcount of academic groups, they were presumably able to share information on a very regular basis, and this surely contributed to their success.
The reliance on CASP dates back to an era when structure prediction did not work at all, and when best practices about data separation and prevention of information leakage were not broadly understood. We exist in a very different climate today. Most researchers understand the issues, and are perfectly capable of constructing training and test sets that properly assess the performance of their methods. My own work in this effort, the ProteinNet dataset, is one concrete contribution I have made to democratize and speed up progress in the field. There will invariably be papers with poor controls and exaggerated claims, but the paranoia of cheating and ineptitude must be balanced with encouraging rigorous but rapidly evolving method development.
CASP serves a crucial purpose, and must continue to do so. DeepMind’s results would not have been nearly as convincing had they not taken place as part of CASP. But we must have a middle ground between the gold standard and a more iterative approach to publication and information exchange. CAMEO helps in this regard, but its targets are often not difficult enough. ProteinNet or something like it, like the NEMO authors’ approach of using CATH-based purging, should be encouraged as a mean to provide acceptable assessment of model quality, especially when it is coupled with release of source code that enables transparent reproduction of the training process.
To be sure, the above will not close the gap between academic and industrial research. There are other, more fundamental problems. For example, competitively-compensated research engineers with software and computer science expertise are almost entirely absent from academic labs, despite the critical role they play in industrial research labs. Much of AlphaFold’s success likely stems from the team’s ability to scale up model training to large systems, which in many ways is primarily a software engineering challenge. While academic labs do not need to perform at the level of Google, they must perform at an adequate enough level to support the core scientific mission of their institutions, and this is not currently happening in my opinion.
An indictment of pharma
What is worse than academic groups getting scooped by DeepMind? The fact that the collective powers of Novartis, Pfizer, etc, with their hundreds of thousands (~million?) of employees, let an industrial lab that is a complete outsider to the field, with virtually no prior molecular sciences experience, come in and thoroughly beat them on a problem that is, quite frankly, of far greater importance to pharmaceuticals than it is to Alphabet. It is an indictment of the laughable “basic research” groups of these companies, which pay lip service to fundamental science but focus myopically on target-driven research that they managed to so badly embarrass themselves in this episode.
If you think I’m being overly dramatic, consider this counterfactual scenario. Take a problem proximal to tech companies’ bottom line, e.g. image recognition or speech, and imagine that no tech company was investing research money into the problem. (IBM alone has been working on speech for decades.) Then imagine that a pharmaceutical company suddenly enters ImageNet and blows the competition out of the water, leaving the academics scratching their heads at what just happened and the tech companies almost unaware it even happened. Does this seem like a realistic scenario? Of course not. It would be absurd. That’s because tech companies have broad research agendas spanning the basic to the applied, while pharmas maintain anemic research groups on their seemingly ever continuing mission to downsize internal research labs while building up sales armies numbering in the tens of thousands of employees.
If you think that image recognition is closer to tech’s bottom line than protein structure is to pharma’s, consider the fact that some pharmaceuticals have internal crystallographic databases that rival or exceed the PDB in size for some protein families.
And if you counter with the argument that machine learning is not pharma’s core expertise, then you only prove my point: why isn’t it? While drug companies wrangle over self-titillating questions like “is AI real?” and “how is deep learning any different than the QSAR we did in the 80s”, Alphabet swoops in and sets up camp right in their backyard. As a result the smartest and most ambitious researchers wanting to work on protein structure will look to DeepMind for opportunities instead of Roche or GSK. This fact should send chills down the spines of pharma executives, but it won’t, because they’re clueless, rudderless, and asleep at the helm.
I am being harsh because this has long been a pet peeve of mine. While companies like Alphabet, Facebook, Microsoft, Intel, and IBM have real research groups with billions of dollars spent on fundamental R&D that has led to Nobel or Turing-grade research, pharmaceuticals engage in “research” so narrowly defined that it rarely contributes to our understanding of basic biology. There is perhaps no better example of this than protein structure prediction, a problem that is very close to these companies’ core interest (along with docking), but on which they have spent virtually no resources. The little research on these problems done at pharmas is almost never methodological in nature, instead being narrowly focused on individual drug discovery programs. While the latter is important and obviously contributes to their bottom line, much like similar research done at tech companies, the lack of broadly minded basic research may have robbed biology of decades of progress, and contributed to the ossification of these companies software and machine learning expertise (there is a reason most newly minted ML PhDs run from pharmas like they’re the plague—they have not cultivated a culture that attracts the world’s best ML talent, in part because of their lack of engagement in basic science.) The AlphaFold episode is only an example of several other problems that have been similarly neglected. It is of course possible that these companies have some newfangled protein structure prediction technology internally, but I’m well networked in these circles and I have seen no indication whatsoever that this is the case.
Smaller and newer companies like AtomWise have done better, focusing more seriously on methodological research, and it will likely take a Silicon Valley-like disruptor to finally turn things around.
The way forward
So what now? Should academics fold up their protein structure research programs and move on to greener and less competitive pastures? And will the space see new entrants from other companies, possibly life science ones? I am still digesting CASP13 and by no means have a definitive recipe, but here are my thoughts so far.
First and foremost, we should recognize what an unqualifiable good thing what just happened is. We, meaning the entire scientific community, have made a major advance on one of the most important problems in biochemistry. Who made the advance is less important than the fact the advance was made, and we should unselfishly rejoice in this fact. I say this cognizant of the fact that my own emotions do not entirely coincide with the sentiment I just espoused, but also cognizant of the fact that we are all adults, and that we must and ought to assess this rationally without letting our tribal affiliations cloud our judgement.
DeepMind’s entry also brings several, unintended benefits. We have a new, world class research team in the field, competitive with the very best existing teams. This has happened maybe once a decade if that. We should welcome them with open arms as, first and foremost, new colleagues with shared purpose. We should encourage them to be as open as the academic teams have been in sharing their research, which they appear to be, and learn from them how to improve our engineering practices, and perhaps more importantly, use their lesson to cultivate a better and more open culture of exchange of ideas, instead of the secretive and siloed behavior that characterizes the field.
DeepMind’s entry also raises the profile of the protein structure problem, likely motivating new students and researchers to work on it, inside of academia and outside. Perhaps DeepMind’s entry will also wake pharmas from their deep slumber, and as a result they too begin to stir with new ideas and resources.
Second, regarding the question of how academic groups should respond scientifically to DeepMind’s entry, I suspect the right answer comes from evolution: adapt. Focus on problems that are less resource intensive, and that require key conceptual breakthroughs and less engineering. Solving protein structure is really multiple problems in one. There is what I would characterize as the canonical problem, the prediction of the overall fold of the native state, and it is the one that most people have focused on including DeepMind. This problem remains unsolved, but it’s clear that for perhaps ~30% of such predictions, we can do very well, and for another ~20% reasonably well. If the trend continues, and there are compelling reasons to argue either way, then something like a solution to this problem is conceivable within ~5 years. That solution may come primarily from better engineering, and so perhaps it represents a less favorable strategic landscape for competition.
Most approaches to the above problem have come from co-evolution based methods and so they are by construction “family-level”. They are able to say a lot less about an individual protein sequence, such as a mutated or de novo designed protein. This is the reason why I have focused on this problem for RGNs, as it is a new frontier. It is unclear if we are even marginally closer to solving this after CASP13—I think there is no indication of any real progress here. And so we could just as well be 20 years out.
Even for MSA / family-level predictions, there is the question of desired accuracy, which hinges on the biological application. If one is predicting protein structures to ascertain their general fold for function classification, then high accuracy is unnecessary. If on the other hand the objective is to design small molecule drugs that bind proteins, which require ~1Å accuracy in the local pocket, it is unclear if we have made any detectable progress.
Finally there is the full realization of the protein folding problem, which concerns not only the final native state but the dynamical trajectory the protein takes to get there, as well as the relative energetics of the near native state ensembles. This is arguably the most important problem for protein function prediction, and it remains very far from being solved.
So let us find and learn the important lessons of CASP13; use it to improve our models and our culture; and, recognizing that we are imperfect, competitive humans, rise above our pettiness to celebrate an important milestone for science.
Post-mortem: RGN @ CASP13
While not the primary subject of this blog post, I would be remiss to not comment on my own participation in CASP13 as a predictor for the very first time! The experience was interesting and informative, but in the end RGNs did not perform well, for reasons I will explain briefly. I should say that it is not possible to know definitively without a thorough analysis, so these are only my best guesses for the moment.
First, given the overall improvement of all co-evolution based methods in CASP13 (even ones that have not changed since CASP12), it appears that the increased availability of protein sequences has widened the gap in information advantage between methods that use co-evolution and those that do not (like RGNs.) I suspect this was the biggest factor in lowering the RGN’s relative ranking. Parenthetically, this suggests a new ultra-hard FM category, single sequence targets without detectable homologs, an idea brought up during CASP13.
Another problem, which I only discovered at the beginning of the CASP13 prediction season, is that all my raw predictions got immediately rejected by CASP’s automatic processing pipeline. This is due to RGN-predicted structures having non-physical torsion angles. In some ways it is unsurprising since, as a machine learning model, RGNs only optimize for what they are trained for, in this case dRMSD. So while the overall global topology of the structures can be quite good, locally the structures are often poor (a point I mention in the paper), and this prevented submissions from going through.
Overlaid backbone traces of experimental (pink) and RGN-predicted (blue) structures for CASP13 FM target T0957s2-D1. While global alignment is good, local alignment is poor, resulting in a low GDT_TS score of 34.
To get around this problem I fed my predicted structures through the Rosetta FastRelax pipeline, which partly defeats the purpose of my method, but my aim was to get structures with sufficiently acceptable local structure to get them past the CASP processing pipeline. This worked most of the time, in the sense that I was able to submit structures, but had the effect of altering them in a way that impacted their accuracy.
It is hard to say yet how much of a contribution this made to reducing RGN performance, and there are other factors that also contributed. For example I used an old RGN model trained on the ProteinNet12 dataset, i.e. couple of years out of date, because I did not have time to retrain for CASP13. I doubt this made a major difference, but it was likely a contributor.
All in all it was a good learning experience, and will give me much to think about over winter break.
Acknowledgments
Thanks to David Baker, Alex Bridgland, Jianlin Cheng, Richard Evans, Tim Green, John Jumper, Daisuke Kihara, John Moult, Sergey Ovchinnikov, Andrew Senior, Jinbo Xu, and Augustin Zidekfor for lively discussions during CASP13 that formed the basis for much of the content of this post.
References
Golkov, V. et al. Protein Contact Prediction from Amino Acid Co-Evolution Using Convolutional Networks for Graph-Valued Images. in Annual Conference on Neural Information Processing Systems (NIPS) (2016).
Jones, D. T., Buchan, D. W. A., Cozzetto, D. & Pontil, M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics28, 184–190 (2012).
Kamisetty, H., Ovchinnikov, S. & Baker, D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. U.S.A. (2013). doi:10.1073/pnas.1314045110
Liu, Y., Palmedo, P., Ye, Q., Berger, B. & Peng, J. Enhancing Evolutionary Couplings with Deep Convolutional Neural Networks. Cell Systems6, 65-74.e3 (2018).
Marks, D. S. et al. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 6, e28766 (2011).
Wang, S., Sun, S., Li, Z., Zhang, R. & Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLOS Computational Biology13, e1005324 (2017).
Xu, J. Distance-based Protein Folding Powered by Deep Learning. bioRxiv 465955 (2018). doi:10.1101/465955
AI, ML, and DataBiologySciencedeep learningprotein foldingprotein structure
For over a decade now I have been working, essentially off the grid, on protein folding. I started thinking about the problem during my undergraduate years and actively working on it from the very beginning of grad school. For about four years, during the late 2000s, I pursued a radically different approach (to what was … Continue reading →
Show full content
For over a decade now I have been working, essentially off the grid, on protein folding. I started thinking about the problem during my undergraduate years and actively working on it from the very beginning of grad school. For about four years, during the late 2000s, I pursued a radically different approach (to what was current then and now) based on ideas from Bayesian nonparametrics. Despite spending a significant fraction of my Ph.D. time on the problem, I made no publishable progress, and ultimately abandoned the approach. When deep learning began to make noise in the machine learning community around 2010, I started thinking about reformulating the core hypothesis underlying my Bayesian nonparametrics approach in a manner that can be cast as end-to-end differentiable, to utilize the emerging machinery of deep learning. Today I am finally ready to start talking about this long journey, beginning with a preprint that went live on bioRxiv yesterday.
What ultimately doomed the Bayesian nonparametrics approach was computational cost—I spent millions of compute hours on the problem without making any headway in getting the models to converge. I thought (and still do) that that formulation captured something fundamental about protein folding, but the mathematical tools for sampling and variational inference were and are not yet mature enough to make it work. Deep learning presented the appealing possibility of casting protein folding as an optimization problem that can be optimized end-to-end using gradient descent. I knew very little about neural networks back then but started studying them seriously in 2012. During the period spanning Sept. 30th, 2014 to Feb. 18th, 2015, I settled on the basic formulation that I would eventually call Recurrent Geometric Networks or RGNs (I track things). It took another three years from conception to realization—deep learning frameworks were nowhere near as mature then as they are now, causing me to start over with a new codebase twice, and RGNs can be quite challenging to train. But, I think they are now finally ready to be released into the wild! For the technical details, go read the preprint. In this blogpost, I’d like to describe the thinking process that led up to them.
From when I first learned about protein folding, and the approaches taken to predict protein structure, I thought it may be possible to predict proteins without conformational sampling and energy minimization, the two pillars of protein structure prediction. The reasons for this have come to underlie what I call the linguistic hypothesis. The basic idea is as follows: there is evidence that today’s proteins emerged out of an ancient peptidic soup, one that may have left its mark on the evolutionary record. I.e., the proteins we see today may in some sense be formed out of primordial peptides. As proteins grew in size and complexity, it would have been advantageous to reuse existing components, to build bigger proteins from existing protein parts. We already know this is true on the level of protein domains, in that larger proteins are often comprised from chaining together smaller globular domains. But the phenomenon of reuse may go further, where even smaller protein fragments (handful of residues to dozens) may reflect an underlying evolutionary pressure to reuse working parts, fragments that fold in tried-and-tested ways (from the perspective of evolution.) If this is the case, then the space of naturally occurring proteins may occupy a very special “manifold”, one that exhibits a hierarchical organization spanning small fragments to entire domains. Other evolutionary pressures could further drive the reuse phenomenon. For example, once a protein-protein or protein-DNA interface is established, presumably through some sort of structural motif, reusing that motif would present an efficient way for the cell to rewire its cellular circuitry. The end result of all this would be the emergence of something resembling a linguistic structure, a grammar that defines the reusable parts and how these parts can be combined to form larger assemblies. Given that this is biology, it’s unlikely to be rigid or minimal. It would be messy and hacky, with many exceptions and ad hoc evolutionary optimizations. But the manifold would be there, potentially discoverable and learnable.
What to me is most exciting about this idea is the emergence of a layer of biological phenomenon that is rooted in physics (obviously), but that can be described independently of it. I.e. the primitives of this phenomenon would be sufficiently abstracted away from the underlying physics that we don’t need physics to model it. We can just operate on the level of protein fragments and motifs, building a probabilistic grammar that describes how these parts combine and interact, without ever resorting to a brute force physics-based simulation. To some this may seem unprincipled. To me, this possibility is more exciting than being able to brute-force simulate protein folding, as such simulations would ultimately only be exercises in doing physics really well and at scale. But the possibility of the existence of a description of protein structure space that can be formulated without resorting to the underlying physics, is the possibility of a theoretical science of proteins, independent of (but ultimately rooted in) physical theory.
My original Bayesian nonparametrics approach very explicitly codified this intuition. That is the advantage (to me) of Bayesian nonparametrics. They are a great way to encode one’s prior about the generative process underlying the phenomenon of interest. In some ways, designing neural network architectures goes in the exact opposite direction. Instead of capturing our prior about the underlying generative process, deep learning works by capturing our prior about the inverse learning process. I.e. the architecture encodes how the phenomenon can best be learned from data. Consider computer vision: images are not actually generated by convolutions, by having copies of cats repeated across our visual field! But, given a natural image, we know that a good prior for learning patterns in images is the imposition of translational invariance. It’s a statement about what makes a good learning process, about regularities in the phenomenon of interest that can be exploited by our learning process.
Recurrent geometric networks try to do this for the linguistic hypothesis I articulated above. There’s nothing in them about ancient peptides or reuse. But they are structured in a way to discover patterns in protein sequences, and to discover hierarchies of such patterns. Perhaps more importantly, they are set up so that the link between sequence and structure is direct and immediate. The signal for whether the learned representation is useful for protein structure prediction comes directly from the predicted structures themselves, because they are explicitly compared to real structures and the deviation between the two is backpropagated to the learned weights of the representation. The hard or what one might call the clever part of RGNs is making this happen, the coupling between the representation and the final output. Ironically, I think the idea itself is very simple and straightforward. I had it over three years ago and it always struck me as very obvious. The real hard part in many ways has been getting it to work.
So are RGNs a panacea? Not at all. This is very much a 1.0 release. They are raw and unpolished. Training them can be quite challenging, like I already mentioned. They do comparatively well on novel protein topologies, but that’s because everyone else does so poorly. They do silly things like predict pretty awful secondary structure, and their predictions can have steric clashes and the like. The specific preprint I just posted has some lame aspects—for example it uses the CASP11 structures as a testbed, instead of the more recent CASP12, for no other reason than the fact that when I first started training them, CASP11 was still current!
But, and this is the key point, I don’t think any of this really matters at this early stage, or distracts from what’s most exciting about them. RGNs can predict protein structures, at a very competitive level, without sampling! Without energy minimization! Without templates! And without the key driver behind the recent successes of protein folding, co-evolutionary data. I was intentionally somewhat puritanical in this paper, in the sense that I didn’t add any bells and whistles such as physical priors, templates, or co-evolutionary information, because I wanted to communicate the key finding that even without all these things, i.e. while being orthogonal to what currently makes protein structure prediction work, RGNs can do pretty well. This means, I am rather certain, that with enough engineering, perhaps Google-scale engineering, it would be possible to make RGNs work really well, maybe shockingly so. To be sure, that’s speculation at this point. But I think it’s clear that they’re a very different way to model protein structure. RGNs reason about proteins in a way distinct from the kind of computations done by molecular dynamics, or fragment assembly, or certainly the sort of optimizations done to extract contact maps from co-evolutionary data. And that to me is fundamentally very exciting.
I hope you take a look and find it as exciting as I do. If I’m successful, this is the beginning of something new, rather than the end of anything. Protein folding has yet to be solved, but we’re living in the most exciting era of this foundational problem; one that may have us see its resolution.
Life, Society, and CultureNIPSRomeUnitedUnited Statesurbanism
Earlier this week I found myself in Rome in the morning with about 20 minutes to spare. Walking around the neighborhood I was staying in (Trastevere), I came across an elderly nun walking along one of the bigger, and more crowded, streets of Rome. As I waited for her to go through a narrow passage … Continue reading →
Show full content
Earlier this week I found myself in Rome in the morning with about 20 minutes to spare. Walking around the neighborhood I was staying in (Trastevere), I came across an elderly nun walking along one of the bigger, and more crowded, streets of Rome. As I waited for her to go through a narrow passage in the sea of people, a young woman pushing a stroller physically nudged her out of the way, using the stroller to deny the physical space in front of and adjacent to the older woman as she overtook her. The nun grimaced but seemed resigned to what happened. I saw this unfold despite having been out for only about ten minutes. In contrast, having walked US streets in San Francisco, Boston, and New York for over twenty years, I don’t recall seeing a similar situation happen even once. It follows that frequentist estimates of such occurrences in American cities and Rome suggest very different underlying distributions.
Not ten seconds passed when I saw yet another very elderly woman, walking at a pace that could not have exceeded a few yards per minute. She was being supported, with interlocked arms, by a much younger woman, possibly her granddaughter, with an apparent infinitude of patience. In the span of the twenty minutes that I walked around, virtually identical scenes unfolded three more times, all with that same infinitude. In contrast, in my over twenty years of walking American cities, I have observed similar scenarios only a handful of times (usually without the patience). Frequentist estimates again suggest very different underlying distributions.
(An important caveat: the age distribution of people in Rome is very different from the aforementioned American cities—far more elderly are visible in Rome. This cuts both ways.)
This prompts me to propose the following Law of Conservation of Empathy: On average (across cultures) the average (across individuals) amount of empathy that an individual displays toward other people is fixed; what changes is how that empathy is distributed across the people that an individual interacts with. To be clear, the capacity for empathy does vary across individuals, but the average capacity of individuals across cultures is largely fixed. I.e. is small, but is high for any given society. More importantly, the point I am making is that how this capacity for empathy is manifested toward people that an individual interacts with varies a great deal between cultures, despite the total amount of it being fixed on average. I.e. is high. Unrelatedly, I think is probably small for most societies. (Sorry I just came back from NIPS.)
In cultures with strong local communities and tribalistic tendencies, most of the (statistically fixed total) capacity for empathy is directed toward family and close friends. If you’re close to someone, they will take good care of you. If you’re not, you’re sort of on your own. Cultures occupying this extreme end of the spectrum are mostly in the Middle East and Asia, are generally what one would think of as being very communal, and tend to exhibit somewhat discourteous behavior toward strangers.
In cultures with strong individualistic tendencies and pluralistic and multicultural outlooks, the capacity for empathy is more uniformly distributed across all persons that an individual interacts with. It doesn’t matter how close you are to someone, they will more or less treat you the same way they would treat a stranger, which is basically with common decency and courtesy, if a little cooly. Cultures occupying this extreme end of the spectrum are mostly in the United States and the English-speaking New World.
European countries generally fall in the middle, as do Latin American ones. In the US, the west coast tilts individualistic, the east coast communal.
This seems consistent with a persistent yet somewhat distressing observation I have made, and that is there appears to be no free lunch when it comes to social interactions. Either people are outwardly nice and fair, but always maintain a certain distance no matter how close you get, or they are a little curmudgeony, but are quite decent once you get to know them. If the empathy pie is truly fixed, then there’s no hope of creating this ideal society I’ve always dreamed of, where people are both outwardly nice and fair, and are capable of very close friendships. Too bad.
(I write the above somewhat in jest. There’s no real evidence backing any of what I’m saying, certainly not in the sense of any real “law” of human behavior.)
AI, ML, and DataDARPAmachine learningPortlandprobabilistic programming
For two weeks last July, I cocooned myself in a hotel in Portland, OR, living and breathing probabilistic programming as a “student” in the probabilistic programming summer school run by DARPA. The school is part of the broader DARPA program on Probabilistic Programming for Advanced Machine Learning (PPAML), which has resulted in a great infusion … Continue reading →
Show full content
For two weeks last July, I cocooned myself in a hotel in Portland, OR, living and breathing probabilistic programming as a “student” in the probabilistic programming summer school run by DARPA. The school is part of the broader DARPA program on Probabilistic Programming for Advanced Machine Learning (PPAML), which has resulted in a great infusion of energy (and funding) into the probabilistic programming space. Last year was the inaugural one for the summer school, one that is meant to introduce and disseminate the languages and tools being developed to the broader scientific and technology communities. The school was graciously hosted by Galois Inc., which did a terrific job of organizing the event. Thankfully, they’re hosting the summer school again this year (there’s still time to apply!), which made me think that now is a good time to reflect on last year’s program and provide a snapshot of the state of the field. I will also take some liberty in prognosticating on the future of this space. Note that I am by no means a probabilistic programming expert, merely a curious outsider with a problem or two to solve.
What is probabilistic programming?
Before I get on with describing my impressions, I will give a very quick primer on probabilistic programming. This is not meant to be in-depth as there are many excellent existing resources. In particular, if you find my discussion too brief then I suggest this introduction, which provides a more substantial treatment. Other accessible resources include an in-depth walk-through using the Church language here, and a portal on probabilistic programming systems (PPSs) here. Incidentally, PPS is the preferred terminology for DARPA, meant I believe to emphasize the fact that PPSs are more than “just” programming languages, as they typically include an inference engine among other things.
At their most basic level, probabilistic programming languages differ from deterministic ones by allowing language primitives to be stochastic. In other words, instead of being restricted to deterministic assignments such as:
x = 5
one can also specify a probability distribution from which x is drawn, e.g.:
x ~ normal(mu = 0, sigma = 1)
Depending on the expressiveness of the language, the distributions from which primitives are drawn can be quite complex (e.g. distributions over functions). The power of probabilistic programming doesn’t come from merely specifying probabilistic primitives however, as that can be easily done with standard programming languages (e.g. rand). Instead it is their ability to condition on observations (or functions thereof). For example, we can write something like the following program:
mu ~ uniform(-10, 10)
x ~ normal(mu, sigma = 1)
observe(x = 5)
The observed value of x is used to constrain the space of possible program executions to a specific subset, namely ones in which x = 5. This in turn makes it possible to perform inference. E.g. we can then add:
infer(mu)
And the PPS will provide an estimated value of mu that corresponds to the conditional distribution of mu given x = 5. Such estimates are typically derived either through sampling approaches like MCMC or through optimization approaches that provide the maximum a posteriori estimate. This makes it possible to construct reasonably complex probabilistic models with relative ease, and in principle, enables machine learning practitioners to explore novel models rapidly. An impressive demonstration of the breadth of models that can be coded using a tiny amount of code can be found here. It includes things like nested Dirichlet processes, infinite probabilistic context-free grammars, and more pedestrian models like Latent Dirichlet Allocation. The “point” of probabilistic programming systems is something that I will expound on below, but it is important to note from the get-go that they are not meant to compete with ML frameworks like scikit-learn. Unlike the latter set, which typically provide a fixed set of models and algorithms, PPSs enable the construction of entirely novel models, and in some cases, inference algorithms. As a natural consequence of this, PPSs are unlikely to be as efficient in running well-trod models as specialized algorithms.
There are many ways to classify and think about all these languages. The space is fragmented right now, with different groups experimenting with widely divergent approaches. The result is that there’s a great deal of diversity and uncertainty, and in some ways this is one of the major challenges facing the field, as I describe later. For now, I found the following criteria to be helpful in thinking about PPSs:
Expressivity
Scalability
Maturity
Programming Paradigm
Use Case / Niche
I will go through each in turn.
Expressivity
How expressive is the language? There are in fact two questions baked into this. One is the theoretical expressivity of the language, i.e. the set of probabilistic programs that it can represent. The second is the practical expressivity of the language, i.e. how easy it is to write programs of increasing complexity. Two languages may in principle be capable of representing any samplable probability distribution, but in practice one language may really be designed for a certain subset of problems, which makes it awkward to express more general distributions. Almost all languages on display were in principle capable of representing arbitrary distributions. The exceptions to this were Dimple, which is restricted to graphical models, specifically factor graphs, and BLOG, which is focused on logic models. Outside of PPAML, Stan and Infer.NET are also languages that are not capable of representing arbitrary probabilistic programs. In some instances claims of expressiveness can be contentious, for example see this blog post on whether Stan is Turing complete. My criteria is based on whether it’s possible to represent arbitrary probability distributions using the language’s built-in constructs.
Venture, Figaro, Church, and Chimple can all in principle represent arbitrary probability distributions. In practice however, the expressivity of these languages varies a great deal. Venture, and its antecedent Church, have the cleanest language design. They were intended for unconstrained probabilistic programming and so their designers took (and are taking, as much of the language design for Venture is still in flux) great care in providing clear and concise semantics for representing probabilistic programs. Figaro and Chimple focus on certain problem domains, for example graphical models in the case of Figaro, which makes them somewhat awkward to use for more general applications. Many of the language’s constructs and general idioms are geared specifically toward graphical models.
Naturally, expressiveness comes at a cost. More expressive languages may not be able to carry out inference as efficiently as the languages with more specialized constructs, since they can’t assume as much about the model. Another potential drawback to expressivity is the ease with which users can write ill-defined probabilistic programs and ones in which efficient sampling is nearly impossible due to the complexity of their resulting models. More expressive languages provide longer proverbial ropes. Finally an issue that all languages face, but in particular the “clean” ones, is the level of abstraction that should be provided to the user. Ideally a handful of powerful constructs would be sufficient to build most probabilistic programs of interest. The design of Venture/Church accomplishes this quite well. For example a Dirichlet process (DP) can be written from scratch in a few lines of code. Unfortunately, such clean implementations written in the language itself end up not being terribly efficient, necessitating that ready-made constructs for commonly used distributions like DPs are built into the language. The need to provide higher-level black box implementations for commonly used distributions detracts from the cleanliness and unification of probabilistic programming. These issues arise in programming languages in general and scientific computing in particular. For example Matlab provides a host of specialized functions for performing linear algebra, and doing the naïve but clean implementation can often prove sub-optimal. These challenging design decisions are some of the more difficult problems facing the PPS ecosystem right now.
Performance
The scale at which PPSs can carry out inference, i.e. the size of data to condition on, and their general performance characteristics was another area in which languages varied considerably. Some, like Church, are focused primarily on language design and the ability to express very complex probabilistic programs (more on this later). While they may not necessarily be inefficient, sampling efficiency is clearly not their priority. Others, like Figaro and the Chimple/Dimple pair, provide efficient sampling for certain subsets of probabilistic programs, for example graphical models. [Ch/D]imple chose an interesting design route where Chimple is a general-purpose PPS that uses Metropolis-Hastings for everything, while Dimple is a separate language that’s meant specifically for graphical models and strives for high performance in that domain.
In the scalability/expressiveness space, Venture is the language that is attempting to have its cake and eat it too, by being simultaneously highly expressive and capable of carrying out serious, industrial-strength, inference. Its approach is two-pronged. First, unlike other languages which only provide one or two sampling techniques, Venture provides a plethora of built-in algorithms, including less common techniques like Slice sampling. Second, Venture exposes the capability to program the inference procedure itself through a built-in inference programming language. For the version at the summer school, what this meant is the ability to tag different random variables as belonging to distinct scopes, and then to define a meta sampling procedure, constructed out of the existing sampling primitives, that specifies how and when the different scopes are sampled. Ultimately their vision is for the inference procedure itself to be an arbitrary probabilistic program.
Figaro also has some inference programming capabilities, although they did not appear to be as extensive as Venture’s, but I suspect this will be an area of future focus for the language. In addition, although Church is not focused on high-performance inference, its programs are converted to JavaScript. This means its performance is actually quite competitive, benefiting from the stupendous efforts that browser developers have put into optimizing compilers.
My impression is that DARPA, and the three highlighted PPSs (Venture, Figaro, and BLOG), all consider high-performance inference to be a major objective. I think all groups are wary of overpromising on this account, as there remain fundamental open questions regarding how doable general inference is, even in principle. Several negative results have already been established about the viability of inference in all samplable distributions, but I suspect as is often the case with such negative results, they are extremely pessimistic and in practice the subset of problems we care about may prove to be much more tractable (the halting problem hasn’t stopped people from building perfectly functional software). For now high-performance inference remains an objective and not a reality, particularly when considering general probabilistic programs. All examples shown were basically toyish in scale, if not in model complexity, and no language currently comes close to competing with custom C/C++ code. For specific subclasses of systems, the situation is different, and languages like Figaro and BLOG can offer competitive performance. This is a fundamental design consideration moving forward, as more expressive languages may end up sacrificing performance because of their inability to assume as much about the underlying set of possible programs.
Maturity
For a field as nascent as probabilistic programming, one might assume that all existing languages are equally (im)mature. This is in fact untrue for several reasons. First, older modeling languages are jumping on the probabilistic programming bandwagon by rebranding themselves. Second, many PPSs started out as tools for constructing graphical models and evolved into more general-purpose modeling languages. Finally, even within the world of genuine probabilistic programming, some languages have had a few years head start on others.
Broadly speaking, the new languages tend to be cleaner and well designed, while the older ones have usually made some questionable design choices. On the other hand, because of the immaturity of the newer languages, they are less optimized from a performance standpoint and suffer from the usual glitches and instabilities of alpha/beta software.
At PPAML the most mature PPS on display was BLOG. There was a competition of sorts (DARPA doesn’t like calling it that) to evaluate the PPSs on “Challenge Problems” that have been designed to test out the expressivity and performance of PPSs. BLOG was the only language able to run the Challenge Problems at the largest scale, including the million birds version of one of the Challenge Problems. This is undoubtedly thanks to the fact that BLOG has been in development for nearly 10 years and is now a well-tested and well-optimized system.
The intermediate case was Figaro, which had more of the trappings of a modern PPS system, including integration into Scala and the expressivity to represent nonparametric distributions, but is perhaps not quite as mature as BLOG given that it’s been in development for only a few years. Venture represented the frontier, a very modern language with substantial potential, but one that is very much a work-in-progress with frequent crashes and spartan support for even basic functional programming constructs. Its developers were forthright about it being alpha software, and I am hopeful that it will rapidly mature over the course of the next year or two.
Since the summer school, Haraku, which was not officially released yet, has since become available. I suspect we will see more languages emerge as well. The fact that there’s such a broad distribution of maturity levels makes choosing a language today somewhat tricky. Investing in a solid language like BLOG or Stan will yield immediate dividends in terms of (relatively) high-performance, reliable environments, and complete documentation. On the other hand, the specter of the new wave of languages arriving soon suggests that committing prematurely to a language now may result in regret down the road. My impression after the summer school is that if one has a “traditional” statistical modeling problem that does not involve nonparametrics, then Stan or BLOG is the way to go. For graphical models, Figaro and other graphical model-specific languages are probably the right approach. For everything else, namely Bayesian nonparametrics, I suggest either taking the plunge now with alpha-level software or waiting until the dust settles.
Programming Paradigm
Like regular programming languages, the question of programming paradigm takes center stage in PPSs. The full zoo of paradigms is already represented by existing PPSs: Figaro is object-oriented, Venture, Church, and Hakaru are functional, while BLOG is logic-based. Having been rescued from the clutches of object-orientation by early exposure to Mathematica (now Wolfram Language), my inclination is strongly functional, and given the mathematical nature of the programs written in PPSs, I think the functional paradigm is particularly well suited. On the other hand, these are modeling languages, and object-orientation can make reasoning about different types of objects and their inter-relationships very natural. I suspect that this difference, perhaps more than any other, will make it possible for multiple languages to survive.
Beyond the programming paradigm, another important issue is whether a PPS exists as an independent programming language or as a domain-specific language (DSL) built on top of a general-purpose language. Here again there is a divide, one that happens to parallel whether the language has academic or industrial roots. Venture/Church, while being dialects of Scheme, are independent languages. Similarly for Haraku and BLOG. Figaro on the other hand is built on top of Scala, while [Ch/D]imple can be used within Matlab or Java.
This is a big distinction and dramatically alters the use cases of the language. The main advantage for independent languages is the cleanliness of language design afforded by the ability to completely rethink what a probabilistic programming language should be. The advantage of DSLs is their ability to leverage the richness of existing libraries of established programming languages. The integration is more than skin deep. Figaro allows any Scala object to be treated probabilistically, enabling some very advanced scenarios, e.g. the ability to reason and manipulate visual graphical objects in a scene probabilistically. The fact that industrial languages have opted for the DSL route is indicative of their more immediate practicality. Venture does try to bridge this gap through tight integration with Python, but its interface is far from seamless.
Use Case / Niche
All the above brings me to what in some sense really matters, and that is the PPSs target use case and niche. Depending on its objective, the capabilities that a PPS must achieve can vary a great deal. Without a clear target market, it is difficult to conjure up the appropriate language design, and to prioritize the features that need to be included.
Many use cases were proffered at the summer school. The biggest and most obvious is for domain experts—engineers, scientists, etc.—to explore and prototype specialized models. This may include machine learning researchers and advanced practitioners. The market for this area is not unlike that for scientific computing platforms such as Mathematica and Matlab, and just like these systems, certain problem sizes may be tractable without having to step outside the PPS.
Another set target specialized domains. Church for example is increasingly focused on cognitive science and psychology, and its team intends to push the boundaries of model expressiveness to capture increasingly subtle models, such as this one. Their focus is not on scale or efficient inference, not even model expressivity per se, but on a certain class of models that make use of self-referential behavior.
Then there are even more specialized use cases, such as the ability to communicate novel models in conference papers using formal probabilistic programs instead of the current mixture of prose, math, and plate notation. Almost all groups mentioned this as a possibility. Naturally the extent to which any language can realistically assume this role will depend a great deal on its readability
Not surprisingly, the focus of any given language tended to correlate with its origin as an academic or commercial project. A language like Figaro, which is designed and commercialized by Charles River Analytics, is putting a strong emphasis on scalability and inference, and is already being used by paying customers. The fact that it may not push the envelope of language design is beside the point. It is also focused on playing nice with existing ecosystems, integrating with Scala and thus Java, and being generally accessible to industry users. Languages with academic origins like Venture are much less concerned, at least currently, on acquiring paying customers and are thus freer to explore bolder language design choices, at the expense of being practical tools that integrate well with a researcher’s existing toolbox.
Challenges
There is a great deal of uncertainty in the field, which presents a challenge in its own way. However, there are also areas of clear difficulty that I see as the key hurdles to be overcome in the coming years.
General Inference
First is the question of how complex a program can get before inference becomes hopeless. We already know that inference in the arbitrarily general case is intractable. The more pertinent question is whether there is a class of problems that is large enough to warrant a programming language to describe it, yet small enough to be amenable to efficient inference. Clearly there are families of programs, for example Bayesian networks or hierarchical DPs (HDPs), that are useful and in which tractable inference is possible. However if the space of such programs is sufficiently fragmented, a high-level language may not be a terribly useful paradigm for specifying probabilistic programs, as they would all be effectively ad hoc and might as well be programmed from the ground up by the researchers who invent them. In some sense, the success of languages like Stan already dispels this notion, as they prove the existence of a useful space of structured probability distributions that can be specified programmatically. But Stan only addresses a small subspace of problems, and in particular for models involving discrete or infinite quantities, this remains very much an open question.
Scalability
Related to general inference but quite distinct from it is the question of scalability. This is not so much about whether a given class of programs is amenable to efficient inference. Rather, assuming a class of problems can be sampled efficiently, can its (generic) description in a programming language be reasoned about in an automated fashion to yield a sampling algorithm that is as efficient (at least asymptotically) as would be designed by a human programmer? Obviously in the limit of AI, the answer is yes, but in the near-term horizon of 1 to 5 years, this will be an important determinant of the success of PPSs. Languages like Figaro don’t really count, because they have specialty constructs that deal with the “design patterns” of probabilistic programming. Similarly my experience with Venture so far made it clear that a naïve generic implementation of an HDP, while correct, will not be sampled from efficiently. The problem is only interesting in the case of programs that are not obviously reducible to an existing construct, and thus the language designers can’t cheat by looking up the right algorithm from a pre-existing library. David Blei’s work on black box variational inference seemed to garner some hope in this area.
Inference Engines
Beyond the above, somewhat theoretical considerations, there is the well-defined task of building the requisite sampling algorithms. Almost all languages provide the basics (MCMC, Gibbs Sampling, etc), but there is a race of sorts to bake in more and more algorithms. Part of the challenge here is not overwhelming the programmer with an overabundance of algorithms, but instead developing intelligent heuristics that remove the guesswork out of this process. I haven’t talked much about Stan (see Bonus), but it is one language that has taken an alternative approach, relying on one sampling approach, Hamiltonian Monte Carlo, to do all the heavy lifting. Such simplicity in design may prove prescient.
Language Design
What a probabilistic programming language should look like may end up being very, very different from what regular programming languages currently look like, and my impression is that few people, if any, really have an idea of how this will settle down. A comment made by one of the senior researchers working on a PPS was telling: he said that in regular programming languages, he can write one hundred lines of code and be fairly confident of its behavior. With a PPS, even ten lines of code can lead to programs whose behavior is entirely unpredictable. This has been my experience as well, and in some ways points to a general drawback of probabilistic programming: it makes things too easy. There is the old adage about certain programming languages handing the programmer enough rope to hang themselves. This phenomenon appears to be widely true of PPSs.
It may in part be due to the prevailing inexperience of all would-be probabilistic programmers, but it’s also clear that PPSs present some fundamentally new challenges. I believe that well-designed languages will provide constructs for concisely writing probabilistic programs in a way that makes it possible to reason about the tradeoffs between complexity and tractability of said programs. Concomitantly, programs should be amenable to automated analysis by compilers, so that general inference becomes possible and the language is not merely reduced to a bag of prepackaged algorithms. Striking the balance between clarity, expressivity, and tractability is a major challenge of this field.
The Future
I will finish by prognosticating on the future of this space. For starters, I do think it is an incredibly exciting area, possibly even the future of machine learning and AI. Deep learning has momentarily sucked the air out of the machine learning room—for good reason—and the slowness and generally poor performance of sampling-based approaches hasn’t helped the cause of probabilistic programming either. But the richness of what can be achieved with probabilistic programming is only beginning to be understood, and if / when major breakthroughs in variational and sampling approaches are made, this area will garner a lot more attention than it currently has. For now, the DARPA infusion seems to have really helped, and the program manager running the PPAML program at the time of the summer school (Kathleen Fisher) appears to have done a great job, from the original solicitation which was incredibly well thought-out to the current organization of the program.
For the near term, my suspicion is that the PPS space will experience a lot of growing pains. The right way forward remains unclear, and I don’t believe anyone has an unassailable vision. This is not a fault, but merely a reflection of the early research state of the field. Too many things, most of which won’t work, still have to be tried. I think that eventually useful PPSs will emerge, but none of the ones in the current crop may end up being the “winner”. Many of the people currently involved in PPSs will likely be the same ones that make it work, but there will be a lot of shuffling and the existing languages will largely disappear. It will take multiple iterations to get this right, as it should.
Horizontal vs. Vertical Integration
One of the keys questions that will have to be addressed is that of horizontal vs. vertical integration. There are two aspects to this. One is whether a PPS is a general programming language that offers everything, including libraries for tooling, visualization, etc, or whether probabilistic constructs are merely added to existing languages. Second is whether a PPS provides the full stack of functionality needed for probabilistic programming, including an inference engine, or whether there will be a decoupling where probabilistic languages specify a language design, and pluggable inference engines can be used with different languages. Not being an expert in this field, my impression is that the jury is out, but there are a lot of subtle interactions between how the language is designed and how inference is carried out that makes this unknown territory. There have been several papers for example showing how probabilistic constructs can be added to regular programming languages, but it is unclear in practice whether this approach will ultimately yield a competitive (performance-wise) and elegant solution.
My suspicion is that there will be room for one or two languages that vertically integrate at least the PPS stack and that truly offer a leap in usability over standard programming languages. There is something to be said for this approach because functional PPS code in a language like Venture is very different from regular programs. A 10-line program can specify a very complex inference task, and likely the longest programs for the foreseeable future will be under 100 lines of code (the situation is different for an OO language like Figaro.) Hence thinking of probabilistic programming as regular programming with probability distributions is misguided. On the other hand, even the best PPSs are unlikely to compete with regular programming languages in terms of libraries and APIs, and so it is likely that they will be made to interface with other languages. More broadly, I expect most PPS solutions to adopt a horizontal approach, focusing on one aspect of the PPS stack. In part this is because designing good sampling algorithms is quite different from designing a good language, and the skills and talents of the people involved tend to be quite different. Furthermore, if more and more regular programming languages acquire probabilistic constructs, the value of plug-and-play inference engines will only increase. The situation may end up being somewhat analogous to functional programming. There are a handful of elegant best-in-class functional languages that are great at what they do and integrate the full vertical stack—Mathematica is my favorite example. On the other hand, functional language constructs have been added and are continuing to be added to a wide array of languages. The winners, at least for the time being, seem to be the ugly jack-of-all-trades. One wrinkle to this discussion is the utility of integrating probabilistic constructs in general programming languages. So far, the applications have been somewhat limited, for example intuitive physics or generative computer vision. The question is whether adding “intelligence” to general purpose languages is something that is broadly useful. If it is, then I think the above considerations apply.
Inference
How to do inference and the relationship of the inference engine vis-à-vis the programming language and the programmer is another interesting question. Incidentally, the PPAML summer school lectures were set up in a way that presumes the programmer does not need to be exposed to the underlying inference engine. I think this was a mistake, given that these are early days, and the students would have benefited from better exposure to the guts of PPS systems. More fundamentally, I suspect that exposing the inference procedure via high-level sampling primitives will be the key to making probabilistic programming work, at least in the short term. There is no free lunch, generally speaking, but what does exist is the possibility of exposing the important knobs to the aspiring probabilistic programmer. What inference programming enables is the ability for a programmer to exploit her understanding of the specifics of her probabilistic program, such as conjugacy relationships, exchangeability properties, and other collapsible aspects of the model, in an easily iterable fashion to try out different sampling strategies. Just as probabilistic programming promises to make the exploration of different probabilistic models accessible, inference programming may make it possible to explore the space of sampling strategies more easily, which in turn can result in efficient sampling procedures of specialized probabilistic programs. This will be especially true if the underlying sampling primitives that a PPS exposes are themselves implemented efficiently. Hoping for anything more, i.e. for a PPS to figure out everything automatically including the most optimal sampling strategy, is probably too much to ask for, for the time being. But if PPSs are able to expose inference programming in an accessible fashion, then they do not need to solve the broader and more difficult problem in a single swoop. They would provide immediate value to statisticians and machine learning practitioners today.
Want To Dabble?
If this discussion got you curious about probabilistic programming, I suggest you give webppl a try, which requires very little upfront investment to get started.
Bonus (Stan)
Stan, the language developed and maintained by Andrew Gelman’s group at Columbia, was not represented at the PPAML summer school. Fortunately I caught a talk about Stan by Bob Carpenter at the Open Machine Learning Workshop at MSR in New York. Here are some of my brief thoughts. I should note that I have not personally used Stan (yet!).
Unlike most languages at PPAML, Stan is increasingly a mature platform used by statisticians and data scientists to do real-world modeling. It is maintained by something like a dozen full-time staff members, and more closely resembles a small professional software team than an academic project run by a handful of graduate students (which is an accurate description of most of the other projects). For practical modeling problems that do not involve discrete variables or variable-sized models, Stan is the PPS to beat. In many ways it has already validated the PPS space by virtue of being widely used in real-world contexts.
The caveat to all the above are the words “discrete” and “variable-sized”. The most glaring omission from the Stan toolkit is support for discrete random variables. Interestingly, at the PPAML summer school this was spun by some of Stan’s competitors as a “philosophical” issue, i.e. that Stan’s creators do not perceive discrete random variables to be a meaningful construct for real-world applications. Bob Carpenter dispelled this notion, stating that it’s simply a practical limitation of the way Stan currently does sampling (presumably due to its reliance on HMC). Unfortunately, all indications point to this being a rather fundamental design limitation, and so I am not holding my breath that the problem will be fixed soon. Stan 3 will not have support for discrete variables.
The lack of support for discrete variables also implies the inability to handle variable-sized models like DPs (this is true for even finite variable-sized models), which are all the rage in Bayesian nonparametrics. Regardless of where one stands on the utility of Bayesian nonparametrics (I think they’re very important), the fact that Stan is unable to address the class of models of most interest to ML researchers means its primary target demographic will be limited to ML practitioners and data scientists. This is of course an important demographic, and is the area where Stan has found a lot of success, but it does limit its potential as a future platform for probabilistic programming. It will be interesting to see if Stan is able to overcome its limitations and become the de facto system, or if one of the new PPAML-sponsored languages can mature enough to become a serious competitor to Stan. Regardless, competition is good for the field, and I look forward to seeing it play out.
Life, Society, and CultureFree SpeechIslamismreligionUnited States
Yesterday’s news about the horrific massacre in Paris shook me really hard. I spent the day very upset, and the night puzzled by my extreme reaction. Terrorist attacks have become fixtures of the daily news, with yesterday alone seeing over a dozen killed in Iraq. Why did this bother me so much? I think I’m … Continue reading →
Show full content
Yesterday’s news about the horrific massacre in Paris shook me really hard. I spent the day very upset, and the night puzzled by my extreme reaction. Terrorist attacks have become fixtures of the daily news, with yesterday alone seeing over a dozen killed in Iraq. Why did this bother me so much?
I think I’m beginning to know the answer. I was born and raised in Iraq under Saddam Hussein, who maintained an iron grip on all forms of communication in the country. No newspaper or TV station or media outlet of any kind was allowed to so much as squeal about the government’s brutality or incompetence. Criticizing Saddam, outside of the tightest and most trusted of family circles, was unthinkable. I developed strong and trusted friendships in elementary school, but never once did the subject of the regime, the most important and profound aspect of our daily lives, ever come up in conversation. And it was common knowledge why. If even one of my friends snitched, everyone else who did not would be in trouble. This was elementary school.
I would often day dream about running up to the roof of our building and shouting at the top of my lungs that I despised Saddam, that I wished him dead. I was 6 or 7 at the time, and that was my day dream.
When I immigrated to the United States, I was skeptical that in any country it would be possible to speak freely. Were people really able to criticize their president? Even call him (or one day her!) names? I was unconvinced, and remained so for years. I would come to discover that there were in fact limits. One cannot incite violence, or knowingly slander a non-public figure. But these were rules that made sense, that enabled as much free speech as possible while protecting everyone’s right to life and liberty.
If there is a single right, a single convenience, a single perk of living in the West that I would not depart with, it would be my right to free speech. Let me repeat this. If there is one single thing I would retain about my life in a democracy, it is the right to free speech. It is that important to me. More than my ability to do research. Or to code. Or do the many other things that define my identity. I would give all those up without giving up my right to free speech.
The reason yesterday’s attacks bothered me so much is because they were directed squarely at me. Je suis Charlie indeed.
I previously blogged on my adventures in self quantification (QS). In that post I wrote about the general system but did not delve into specific projects. Ultimately however the utility of self quantification is in the detailed insights it gives, and so I’m going to dive deeper into a project that passed a major milestone earlier today: publication of a paper. If … Continue reading →
Show full content
I previously blogged on my adventures in self quantification (QS). In that post I wrote about the general system but did not delve into specific projects. Ultimately however the utility of self quantification is in the detailed insights it gives, and so I’m going to dive deeper into a project that passed a major milestone earlier today: publication of a paper. If you’re interested in the science behind this project, see my other post, A New Way to Read the Genome. Here I will focus on the application and utility of QS as applied to individual projects.
I have been working on this project in some capacity for about two years. The first thing I wondered about is how much time in total it has consumed. The answer is 1,363 hours and 45 minutes, or about 34 standard work weeks. These numbers reflect actual worked time; things like goofing off, taking bathroom breaks, and chatting with coworkers are excluded. All supporting activities that are not specific to this project, for example attending group meetings and scientific lectures, are also excluded, and so in reality the total time consumed was much more. In addition, the time and effort put in by my collaborators is not accounted for. Given the final results, I’m fairly happy with how my time was spent. The ability to make such an assessment is one of the biggest advantages of self quantification. I don’t have to wonder whether I’m spending my time right, nor do I have to fret about all the wasted hours. I can make more informed judgements. How long did the “SH2 project” take? 1,363 hours.
Of course it gets more interesting when we dig deeper. The first thing I wondered about is task breakdown. What did I actually spend my time on? Here it is:
Not surprisingly, and somewhat reassuringly, most of my time was spent on research. This means either writing code, working out a model, or just thinking. It excludes the consumption of other people’s work and so in some sense captures my net useful output. The fact that it is my top activity for this project is good news, but it barely crosses the 50% threshold. Instead, and this came as an absolute shock, almost 30% of the time was spent “disseminating”! That mostly refers to writing the actual paper, but also includes giving (and preparing) talks, and all sorts of outreach. The fact that it consumed so much is a little disconcerting, and suggests, for me at least, that writing a paper is a major commitment. It is a significant part of the project and should not be taken lightly. In particular, this argues against publishing “me too” papers (to be sure, the fact that this paper landed in a good journal raised the bar for quality and polish.)
If I were to tease out the one major insight that’s come from this QS analysis it would be the time spent on writing; it is a very non-trivial piece of information. But there are others. Somewhat surprisingly, this paper didn’t require all that much literature reading. This is in part because it was in a space that I’m intimately familiar with and so I didn’t need to learn a lot for it, in contrast to another project i’m working on (see below). It also didn’t require much in the way of planning and strategizing, largely because I walked in knowing what needed to be done (again see below). On the other hand, the administrative burden was quite high. Over 10% of my time was spent on logistics, mostly meetings. This is to be expected given the collaborative nature of the project, and all in all is actually much lower than I feared. The 10% was well worth it, and knowing that it was “only” 10% has reassured me of the worth of such collaborative projects.
Just for fun, by breaking things down over time, I get this:
The massive spike corresponds to the major push for writing the paper. The colors reflect that too, where research activity plunged and writing soared. I should note that I did not magically triple my output during that period. The spike mostly represents a shift in priorities, when all other projects were put on the back burner. There was a phase during which I increased my total output over baseline by 42%, but only for about 4 weeks or so. Needless to say I had a very “minimal” lifestyle during that period. In fact I even stopped going to work to save on commuting time.
The time series analysis doesn’t provide any insights that I didn’t already know, but it is gratifying to see it all in one place. What is more interesting is comparing this project to another, riskier project, on which I have nothing published yet. The second project is very different, taking on a virtually impossible problem using a completely untested approach. While I didn’t quite know it beforehand, these differences are starkly reflected in the activity breakdown:
Research still dominates, but less so than before. What is completely different however are all the other pieces. Relatively little time is spent on logistics or writing, reflecting the fact that I’m working on the project alone. Moreover, an enormous amount of time is spent on reading the literature and on planning and strategizing, a full 50% in fact. This is a vastly different situation from the first project, due to the emphasis on execution vs. fundamental research. In some ways however this is also suggesting that the second project is not proceeding so well, that my time allocation is off.
The time series here is actually more interesting:
The yellow spikes refer to major learning efforts, followed by major bursts of research, bust, and then repeat. The lull in the middle largely coincides with the surge in the other project. Nonetheless, knowing what I know about the project, it is clear to me that I am spending far more time than I should be learning and strategizing, and this too is very actionable information. The temporal dynamics are useful because they point to a pattern in my approach that is suboptimal, where I alternate between exclusive modes of learning and doing. More recently I have tried a more integrated approach, and that appears to be reflected in recent months.
On the whole I hope this analysis provides a window into how QS can be practically useful. After my original post I received many comments questioning the value of such a system, which was puzzling to me as I tried to emphasize its utility by highlighting actionable insights. Hopefully I made a more convincing case this time around. If you have any questions feel free to leave them in the comments.
Update (11/14/14): A reader (mokestrel) asked in the comments below about the distribution of contiguous time blocks spent on writing vs. other tasks. This is an excellent question as writing does often require long blocks of uninterrupted attention, and one would expect to see this in the quantitative data. The first thing I did was just to plot a histogram of the lengths of uninterrupted writing blocks vs. all other tasks for the same project (x-axis is hours).
The distribution is shifted to the right, although it’s not a dramatic effect. I realized however that very short breaks often divide what are virtually contiguous blocks, and so it makes sense to fuse blocks that are not separated by anything other than a break. Here’s the adjusted distribution:
Now the effect is much more dramatic, and there are in fact chunks of time that are longer than 12 hours, corresponding to days where all I did was write! I recall a period of about one week where that was more or less true, and the above outliers likely reflect it. Now I know what to strive for next time I write…
I am pleased to announce that earlier today the embargo was lifted on our most recent paper. This work represents the culmination of over two years of effort by my collaborators and I. You can find the official version on the Nature Genetics website here, and the freely available ReadCube version here. In this post, I … Continue reading →
Show full content
I am pleased to announce that earlier today the embargo was lifted on our most recent paper. This work represents the culmination of over two years of effort by my collaborators and I. You can find the official version on the Nature Genetics website here, and the freely available ReadCube version here. In this post, I will focus on making the science accessible to the lay reader. I have also written another post, The Quantified Anatomy of a Paper, which delves into the quantified-self analytics of this project.
We set out to address an easy-to-state but difficult-to-solve problem: predict, from genetic sequence alone, the consequences of mutations. This is a fundamental problem that lies at the heart of genomics, as our ability to obtain data continues to far outpace our ability to make sense of it. We still cannot, in the general case, understand what any given mutation does. But the work we published today makes a small step in this direction. For a subset of proteins involved in cellular signaling, we are now able to predict how any single mutation affects their ability to interact with their partner proteins. This means that for all diseases effected by mutations—we focus on cancer—we can examine how signaling pathways are rewired in the disease state. This can lead to a better understanding of the basic biology of signaling in healthy and diseased cells, and to the development of drugs that target previously unknown proteins in signaling pathways.
Statistical Mechanics By Way Of Machine Learning
The basic idea behind our model is to cast a statistical mechanical model into a machine learning formulation. It is a well-observed fact of machine learning that the model formulation, what the inputs and outputs are and how the data is represented, often matter far more than the specific choice of algorithm used. I believe this was particularly true in our case and explains to some extent why it was possible to make substantial progress on this relatively well-studied problem.
The main issue with existing methods is that they largely fall into two camps, neither of which optimizes the complexity/statistical power trade-off very well. The first camp comprises general protein-protein interaction methods. Such methods can in principle predict the binding affinity of any two proteins, but in practice they are limited to qualitative predictions. General methods take on a significantly more challenging problem than the single domain problem, because arbitrary proteins can bind other proteins in effectively arbitrary ways. This would not be catastrophic if it weren’t for the fact that the number of new data points gained, by pooling all existing data on all types of protein-protein interactions, is far from sufficient to offset the gain in model complexity. Hence general protein-protein interaction methods fall on the too little data for too much model complexity spectrum of models. The other camp encompasses domain-specific methods. The definition of what constitutes a “domain” varies, but for these methods, it typically refers to an individual protein domain, e.g. one of the 100+ SH2 domains in the human proteome. Each model is specific to a single domain. This results in low model complexity (relative to the general protein-protein interaction case), since a given domain is unlikely to vary all that much between its interaction partners, but also equally low statistical power, since only data that is specific for the individual domain is used. Consequently while these approaches solve the model complexity problem, they also lose the data richness of general protein-protein interaction methods.
The key to solving the SH2 domain problem, or any machine learning problem, is to optimize the trade-off between model complexity and statistical power. Unlike existing approaches, our model was not domain-specific, but generally applicable to any SH2 domain. This had the benefit of making it possible to model mutations in SH2 domains, something that was not possible before. Furthermore, given that most SH2 domains vary little between one another, no substantial increase in complexity was incurred by generalizing our model to any SH2 domain sequence. On the other hand, by pooling data from all SH2 domains, we effectively gained two orders of magnitude of additional data for what I suspect is a nominal increase in model complexity. Hitting this sweet spot of model complexity vs. statistical power is one of the key enabling aspects of our model.
The approach we took in tackling this problem synthesizes structural biology, the study of the shape and motion of individual biological molecules, with systems biology, the study of how multiple biological molecules assemble to form functioning systems that carry out cellular growth, division, motion, signaling, and myriad other tasks. To make a tractable first step, we focused on a single family of proteins, ones containing what are known as Src Homology 2 (SH2) domains. Loosely speaking, domains are parts of proteins that function independently and are reused repeatedly throughout evolution in many proteins. SH2 domains in particular are critical to cellular signaling. When the cell senses an external stimulus, information encoding that stimulus is propagated throughout the cell through the use, in part, of SH2 domains that interact with other proteins to form a chain of signaling events, passing information from one molecule to another.
We began by building a model of how individual SH2 domains interact with their protein partners. Such models had been built before, but they lacked the necessary precision to distinguish between two proteins that differed by only a single residue. Making progress in this instance required that we step back and think about the formulation of the problem, how it is typically stated and how it may be restated, before barging ahead with new algorithms. While it is often difficult to pinpoint the precise reason why a new model works where previous ones did not, I do suspect that this reformulation was the key ingredient. For more on the technical details see the box “Statistical Mechanics By Way Of Machine Learning”.
Once we had a working model of individual SH2 domains, we set out to test it computationally and experimentally. The model made a number of surprising predictions, particularly for a protein about which little was known before, by quadrupling the number of its interaction partners. When the initial experimental results came back positive, we were ecstatic, as the agreement was not only qualitative but quantitative. It seemed like it may be possible to model SH2 domains after all.
The next step proved particularly challenging however. Thus far we had been modeling individual SH2 domains, but real proteins are comprised of multiple domains, sometimes dozens. To make useful predictions about the effects of mutations we needed to model proteins in their entirety. This was unexplored territory as existing models focused on individual domains. We would start down a path, often arriving at what seemed like a solution only to discover some critical flaw. For me personally, this work had a distinctly theoretical flavor, very different from the usual machine learning to which I am accustomed. Eventually, after several false starts, we arrived at we believe is the right conceptual model. It solves several problems at once, including the ability to quantify, in an interpretable way, the likelihood that a mutation will lead to a change in protein function that is consequential, i.e. detectable and biologically meaningful. See the box “Deriving an Interpretable Metric” for more.
Armed with this new model, we were now in a position to analyze the effects of mutations in a given disease on SH2 signaling in humans. We decided to focus on cancer because of the inherent relevance of the problem, as cancer is known to impact signaling pathways. Thanks to large-scale publicly funded efforts, thousands of tissue samples from cancer patients have already been taken and their genomes sequenced. This enabled us to analyze the effects of these cancer mutations on the SH2 signaling network. One of the first things to emerge was that cancer mutations seem to target connected subnetworks in the larger human SH2 network. Below I show a figure for kidney cancer which illustrates this.
Individual proteins are shown as nodes, with edges between nodes indicating affinity for interaction. Edges that are perturbed in kidney cancer are shown in green and orange. A priori, there is no reason for the perturbed edges to “cluster” the way they do, i.e. for them to form a connected subnetwork within the larger network. But they repeatedly seem to do this, with different subnetworks targeted in different types of tissue. These subnetworks are suggestive of signaling chains that play a role in cancer function. In any given patient, only one of these edges may be disrupted, and that disruption may be sufficient to dysregulate the entire chain. If one were to examine one patient at a time, these subnetworks would not have emerged. By pooling mutations from multiple patients however, one is able to observe the extent and connectivity of these potential signaling chains.
Deriving an Interpretable Metric
One of the more interesting, and challenging, aspects of the project was deriving a quantity to denote the importance of a mutation. While it’s easy to construct an ad hoc metric, we wanted a quantity that is interpretable, that does not require mental gymnastics to understand. We also wanted this quantity to be biologically relevant, to correspond to changes that would be consequential and detectable experimentally. With respect to interpretability, one challenge was the fact that we had two probabilities to contend with, corresponding to the likelihood of an interaction before and after a mutation. A simple ratio (or difference) of probabilities, while an easy choice, does not have an obvious physical meaning. Furthermore, a ratio would mask important aspects of the change in binding affinity. For example, a strong interaction becoming a very strong interaction would register the same as a non-existent interaction becoming a strong interaction.
With respect to biological relevance, we needed to contend with the fact that mutations occur in domains residing in complex protein “contexts”, i.e. proteins comprised of other domains and binding sites, all of which have the potential to interact with one another. From a biological standpoint, disruption of a single domain-domain interaction may not be consequential or even experimentally detectable. Furthermore, the natural variation in instances of interactions between proteins meant that sometimes two proteins may register as interacting and sometimes as not merely due to noise.
The quantity we ultimately derived, termed , addresses all these issues, and is, mathematically, a probability in the formal sense. Intuitively, is the probability that a given interaction between two proteins will be qualitatively altered, i.e. in an experimentally detectable way, in a given disease. Technically, we first derive the probability of a hypothetical “double experiment” in which the state of a protein complex (bound vs. unbound) is simultaneously measured before and after a mutation. The set of all possible outcomes of these double experiments constitute what is known as a canonical ensemble. We consider the subset of states in this ensemble in which the pre- and post-mutation states differ and in which the change is localized to the mutated site. We compute the probability of this subset of the ensemble, and then take the expectation of this probability over all possible mutations in a given disease, estimated empirically. This expectation is the value of .
In addition to providing intuitively interpretable semantics, also proves to be very useful. As described in the paper, ranking genes using this quantity allows us to fish out proteins involved in cancer which may serve as targets for therapeutic interventions.
On the “practical” side, the method also seems to do something rather useful: find cancer proteins. Such proteins have the potential to serve as targets for drugs. Although cancer genome data provide information on which genes are mutated, most such mutations are “passengers”, there for the ride but inconsequential for cellular function. Mutations predicted by our model to be consequential were overwhelmingly more likely to occur in proteins already known to be cancer-causing or cancer-suppressing than other cancer mutations.
The model also sheds light on a sort of “dark matter” of genetic mutations, ones that are very infrequent and possibly patient-specific and thus nearly impossible to detect using statistical analysis alone. By analyzing this dark matter of genetic mutations, we discovered that on average, they are as likely as recurring mutations to cause disruptions in signaling networks. This suggests that they are severely understudied and must be examined using models that directly predict the functional consequences of mutations.
Above and beyond the specific findings, this work is one of the opening rounds in what I suspect will be a process of synthesis for structural and systems biology. As I mentioned at the beginning, structural biology studies the very small, the building blocks of all biological systems, while systems biology aims to make sense of the interactions of these building blocks. Although it may seem natural to marry the two, historically this has been difficult, as structural biology is primarily applied to individual molecules, making it difficult to scale across whole systems. On the other hand, systems biology models incorporate many components, but do so in a coarse-grained fashion that ignores most quantitative details. Such an approach can often yield very useful insights, but in biology, the devil is very much in the details, and a synthesis of the precision of structural biology with the broadness of systems biology may open entirely new vistas.
This work represents a structural approach to systems biology because it uses structural information to build a model of SH2 domains that is then applied systems-wide, making predictions for all proteins involved in this signaling system. It is also a systems approach to structural biology because it uses what is known about the local neighborhood of a protein, i.e. its interaction partners in the network, to determine whether a mutation will have consequential effects on signaling. The system provides the context in which mutations occur, and this information is exploited.
So what next? Soon after starting graduate school, I found myself asking the question: if I knew the interaction partners of every protein in the cell, what would I do with that information? I didn’t have a good answer then, and I don’t have a good answer now. Back then however, the question was premature. Now I am no longer so sure.
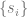

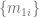
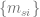




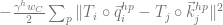










![\text{Var}_{\text{societies}}\left[E_{\text{individuals}}[\text{empathy}]\right]](https://s0.wp.com/latex.php?latex=%5Ctext%7BVar%7D_%7B%5Ctext%7Bsocieties%7D%7D%5Cleft%5BE_%7B%5Ctext%7Bindividuals%7D%7D%5B%5Ctext%7Bempathy%7D%5D%5Cright%5D&bg=ffffff&fg=666666&s=0&c=20201002) is small, but
is small, but ![\text{Var}_{\text{individuals}}[\text{empathy}]](https://s0.wp.com/latex.php?latex=%5Ctext%7BVar%7D_%7B%5Ctext%7Bindividuals%7D%7D%5B%5Ctext%7Bempathy%7D%5D&bg=ffffff&fg=666666&s=0&c=20201002) is high for any given society. More importantly, the point I am making is that how this capacity for empathy is manifested toward people that an individual interacts with varies a great deal between cultures, despite the total amount of it being fixed on average. I.e.
is high for any given society. More importantly, the point I am making is that how this capacity for empathy is manifested toward people that an individual interacts with varies a great deal between cultures, despite the total amount of it being fixed on average. I.e. ![E_{x,y\in \text{societies}}\left[D_{\text{KL}}\left[\text{Dist}_{\text{towards\ others}}^{(x)}[\text{empathy}]\parallel \text{Dist}_{\text{towards\ others}}^{(y)}[\text{empathy}]\right]\right]](https://s0.wp.com/latex.php?latex=E_%7Bx%2Cy%5Cin+%5Ctext%7Bsocieties%7D%7D%5Cleft%5BD_%7B%5Ctext%7BKL%7D%7D%5Cleft%5B%5Ctext%7BDist%7D_%7B%5Ctext%7Btowards%5C+others%7D%7D%5E%7B%28x%29%7D%5B%5Ctext%7Bempathy%7D%5D%5Cparallel+%5Ctext%7BDist%7D_%7B%5Ctext%7Btowards%5C+others%7D%7D%5E%7B%28y%29%7D%5B%5Ctext%7Bempathy%7D%5D%5Cright%5D%5Cright%5D&bg=ffffff&fg=666666&s=0&c=20201002) is high. Unrelatedly, I think
is high. Unrelatedly, I think ![E_{x,y\in \text{individuals}}\left[D_{\text{KL}}\left[\text{Dist}_{\text{towards\ others}}^{(x)}[\text{empathy}]\parallel \text{Dist}_{\text{towards\ others}}^{(y)}[\text{empathy}]\right]\right]](https://s0.wp.com/latex.php?latex=E_%7Bx%2Cy%5Cin+%5Ctext%7Bindividuals%7D%7D%5Cleft%5BD_%7B%5Ctext%7BKL%7D%7D%5Cleft%5B%5Ctext%7BDist%7D_%7B%5Ctext%7Btowards%5C+others%7D%7D%5E%7B%28x%29%7D%5B%5Ctext%7Bempathy%7D%5D%5Cparallel+%5Ctext%7BDist%7D_%7B%5Ctext%7Btowards%5C+others%7D%7D%5E%7B%28y%29%7D%5B%5Ctext%7Bempathy%7D%5D%5Cright%5D%5Cright%5D&bg=ffffff&fg=666666&s=0&c=20201002) is probably small for most societies. (Sorry I just came back from
is probably small for most societies. (Sorry I just came back from 






 , addresses all these issues, and is, mathematically, a probability in the formal sense. Intuitively,
, addresses all these issues, and is, mathematically, a probability in the formal sense. Intuitively,